
Harmony Biosciences, LLC (Harmony) announced the U.S. Food and Drug Administration (FDA) approval of Wakix (pitolisant) for the treatment of excessive daytime sleepiness (EDS) in adult patients with narcolepsy. Wakix is the first and only treatment approved for patients with narcolepsy that is not scheduled as a controlled substance by the U.S. Drug Enforcement Administration (DEA).
“We are extremely proud to bring Wakix to market for those living with narcolepsy, a chronic, debilitating, rare neurologic disorder,” said Harmony’s Chairman and Chief Executive Officer, John C. Jacobs. “At Harmony we share a vision to develop novel treatment options for people living with rare diseases, with a focus on those that affect the central nervous system. The approval of Wakix strengthens our commitment to making that vision a reality.”
Wakix, a first-in-class medication, is a selective histamine 3 (H₃) receptor antagonist/inverse agonist that works through a novel mechanism of action to increase the synthesis and release of histamine, a wake-promoting neurotransmitter in the brain. Wakix is administered orally once daily in the morning upon wakening.
“The approval of Wakix provides healthcare professionals managing people living with narcolepsy a new and important treatment option for their patients,” said Harmony’s Chief Medical Officer, Jeffrey Dayno, M.D. “Additionally, Wakix is the only non-scheduled treatment option approved for adult patients with narcolepsy, and it offers an important benefit/risk profile to address the unmet medical need that exists in people living with narcolepsy.”
The efficacy of Wakix for the treatment of EDS in adult patients with narcolepsy was evaluated in two multicenter, randomized, double-blind, placebo-controlled studies (HARMONY 1 and HARMONY 1bis). These studies included a total of 261 patients who were randomized to receive Wakix, placebo, or active control; these patients had a median age of 37 (HARMONY 1) and 40 (HARMONY 1bis). Treatment duration was eight weeks, with a three-week dose titration phase followed by a 5-week stable dose phase; 75% to 80% of the patients in these studies had a history of cataplexy. In both of these studies, WAKIX demonstrated a statistically significant improvement in EDS as measured by the Epworth Sleepiness Scale (ESS) score. In the placebo-controlled trials conducted in patients with narcolepsy with or without cataplexy, the most common adverse reactions (occurring in ≥5% of patients and at twice the rate of placebo) with the use of WAKIX were insomnia (6%), nausea (6%), and anxiety (5%).
Wakix will be commercially available to healthcare professionals and appropriate patients in the U.S. in the fourth quarter of 2019.
About Wakix (pitolisant)
Wakix is a first-in-class medication approved for use in the U.S. for the treatment of excessive daytime sleepiness in adult patients with narcolepsy. It was granted orphan drug designation for the treatment of narcolepsy in 2010. WAKIX is a selective histamine 3 (H₃) receptor antagonist/inverse agonist. The mechanism of action of Wakix is unclear; however, its efficacy could be mediated through its activity at H₃ receptors, thereby increasing the synthesis and release of histamine, a wake promoting neurotransmitter. WAKIX was designed and developed by Bioprojet Societe Civile de Recherche (Bioprojet), who has marketed the product in Europe since its approval by the European Medicines Agency in 2016. Harmony has an exclusive license from Bioprojet to develop, manufacture and commercialize pitolisant in the United States.
INDICATION AND USAGE
Wakix (pitolisant) is approved for the treatment of excessive daytime sleepiness (EDS) in adult patients with narcolepsy.
IMPORTANT SAFETY INFORMATION
Do not use Wakix if you have severe liver disease.
Tell your healthcare provider about all your medical conditions, including if you have heart rhythm irregularities, were born with a heart condition, or the levels of electrolytes in your blood are too high or too low. Wakix has an effect on the electrical activity of the heart known as QT/QTc prolongation. Medicines with this effect can lead to disturbances in heart rhythm, which are more likely in patients with risk factors such as certain heart conditions, or when taken in combination with other interacting medicines. Tell your healthcare provider about all the other medicines you take.
The risk of QT prolongation may be greater in patients with liver or kidney disease. Wakix is not recommended in patients with end stage kidney disease.
The most common side effects seen with Wakix were insomnia, nausea and anxiety. Other side effects included headache, upper respiratory infection, musculoskeletal pain, heart rate increased, and decreased appetite. These are not all the possible side effects of Wakix. Tell your healthcare provider about any side effect that bothers you or that does not go away.
Tell your healthcare provider about all the medicines you take or plan to take, including prescription and over-the-counter medicines. Some medicines can increase the amount of Wakix that gets into your blood and some medicines can decrease the amount of Wakix that gets into your blood. The dosage of Wakix may need to be adjusted if you are taking these medicines.
Wakix can also decrease the effectiveness of some medicines, including hormonal birth control methods. You should use an alternative non-hormonal birth control method during treatment with Wakix and for at least 21 days after discontinuation of treatment.
Tell your healthcare provider if you are pregnant or planning to become pregnant. There is a pregnancy exposure registry that monitors pregnancy outcomes in women who are exposed to Wakix during pregnancy. You are encouraged to enroll in the Wakix pregnancy registry if you become pregnant while taking WAKIX. To enroll or obtain information from the registry, call 1-800-833-7460.
The safety and effectiveness of Wakix have not been established in patients less than 18 years of age.
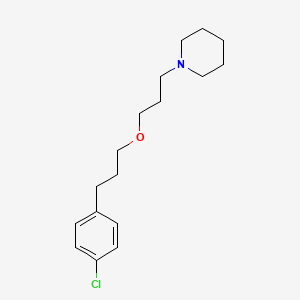
https://pubchem.ncbi.nlm.nih.gov/compound/Pitolisant#section=2D-Structure

 Pibrentasvir
Pibrentasvir
 Saxagliptin and
Saxagliptin and  (Metformin)
(Metformin)



