
Boehringer Ingelheim’s Hernexeos (zongertinib tablets) has been approved by the U.S. Food and Drug Administration (FDA). The kinase inhibitor is indicated for the treatment of adult patients with unresectable or metastatic non-squamous non-small cell lung cancer (NSCLC) whose tumors have HER2 (ERBB2) tyrosine kinase domain activating mutations, as detected by an FDA-approved test, and who have received prior systemic therapy.1

This indication is approved under accelerated approval based on objective response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.1
“With the approval of zongertinib, we have an effective, targeted, orally administered treatment option for patients with HER2 (ERBB2)-mutant non-small cell lung cancer in the U.S. that not only elicits a durable response but, importantly, has a manageable safety profile,” said Dr. John Heymach, MD, PhD, chair of Thoracic/Head and Neck Medical Oncology at The University of Texas MD Anderson Cancer Center, and coordinating investigator for the Beamion-LUNG 1 trial. “In a patient population where there are currently limited treatment options, this approval represents a significant advancement in cancer care.”
Accelerated approval is based on data from the Phase Ib Beamion-LUNG 1 trial, demonstrating an objective response rate of 75% (N=71), 6% of patients had a complete response and 69% of patients had a partial response and a duration of response of ≥6 months in 58% of patients (n=53).1 Positive results from the Beamion-LUNG 1 trial were previously presented at the American Association for Cancer Research (AACR) Annual Meeting 2025 and simultaneously published in The New England Journal of Medicine.2
Zongertinib demonstrated a manageable safety profile with a 2.9% discontinuation rate. In the pooled safety population, the most common (> 20%) adverse reactions were diarrhea (53%), hepatotoxicity (27%), rash (27%), fatigue (22%), and nausea (21%).1
“We are grateful to be able to bring forward Hernexeos, which has the potential to reset the benchmark for those living with HER2-mutant advanced non-small cell lung cancer, a condition associated with a particularly poor prognosis,” said Shashank Deshpande, Chairman of the Board of Managing Directors and Head of Human Pharma at Boehringer Ingelheim. “Believing in the power of scientific innovation, we aim to provide meaningful improvements to this patient population. Recognizing its potential, we accelerated development to deliver this new treatment option to patients within four years of starting the first clinical trial."
Harnessing the power of precision medicine by targeting HER2 mutations in lung cancer
HER2 (ERBB2) mutations occur in approximately 2–4% of NSCLC cases and are associated with a poor prognosis and higher incidence of brain metastases.3,4,5Alterations in the HER2 (ERBB2) gene, including mutations, amplification and overexpression, trigger uncontrolled cell proliferation, inhibiting cell death, and promoting tumor growth and spread.3,5 Comprehensive biomarker testing using next generation sequencing determines a patient’s eligibility for treatment with zongertinib by identifying HER2 (ERBB2)-mutant advanced NSCLC.1,5
“The advocacy community is thrilled about the approval of zongertinib, as it is another testament to the importance of personalized options for lung cancer that allow for a much more targeted approach for a subgroup of patients who have been waiting many years for innovative treatments,” said Marcia Horn, President and CEO, International Cancer Advocacy Network and Executive Director of the Exon 20 Group/HER2 Warriors. “Understanding your cancer’s unique biomarkers, including HER2, through comprehensive testing is critical for all patients with NSCLC, as it can unlock targeted treatment options.”
About non-small cell lung cancer (NSCLC)
Lung cancer claims more lives than any other cancer type and the incidence is set to increase to over 3 million cases worldwide by 2040.5,6 NSCLC is the most common type of lung cancer.3 Due to a lack of symptoms and misdiagnoses,7 most patients with NSCLC present at stage III or IV, where the disease has metastasized locally or to other organs.8 The estimated 5-year survival rate historically has been less than 10% for metastatic disease.9,10,11 People living with advanced NSCLC can experience a detrimental physical, psychological, and emotional impact on their daily lives.12,13,14
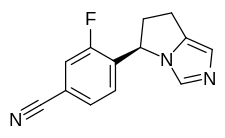
About zongertinib
Zongertinib is a tyrosine kinase inhibitor (TKI) that selectively inhibits HER2 (ERBB2).1 This orally administered, targeted therapy was approved as Hernexeos® (zongertinib tablets) under the FDA’s Accelerated Approval Program, after securing Priority Review as well as Breakthrough Therapy and Fast Track Designations.1
The treatment is being evaluated in ongoing trials, across a range of advanced solid tumors with HER2 alterations.
https://en.wikipedia.org/wiki/Zongertinib#:~:text=Society%20and%20culture-,Legal%20status,under%20the%20brand%20name%20Hernexeos.








 Pibrentasvir
Pibrentasvir
 Saxagliptin and
Saxagliptin and  (Metformin)
(Metformin)