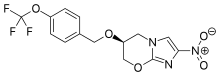
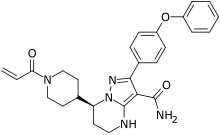
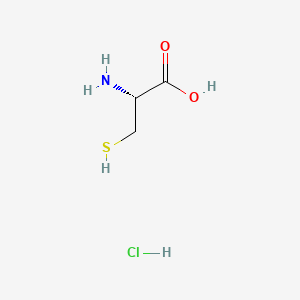
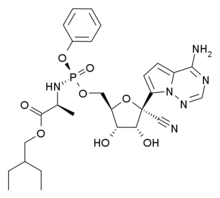
In continuation of my update on Luspatercept
Celgene Corporation (NASDAQ: CELG) and Acceleron Pharma Inc. (NASDAQ: XLRN) today announced the U.S. Food and Drug Administration (FDA) has approved Reblozyl (luspatercept-aamt) for the treatment of anemia in adult patients with beta thalassemia who require regular red blood cell (RBC) transfusions. Reblozyl is not indicated for use as a substitute for RBC transfusions in patients who require immediate correction of anemia. Reblozyl is the first and only FDA-approved erythroid maturation agent, representing a new class of therapy which works by regulating late-stage red blood cell maturation to help patients reduce their RBC transfusion burden.
“Today’s approval is an important milestone and underscores our continued commitment to patients with hematology disorders,” said Nadim Ahmed, President, Global Hematology and Oncology for Celgene. “There are very limited options for patients living with anemia due to beta thalassemia who are dependent on long term red blood cell transfusions. We are pleased to make Reblozyl available as a new therapy for these patients to help address their anemia, a significant clinical complication of beta thalassemia.”
“We’re thrilled that Acceleron’s first approved medicine is one with the potential to help patients with beta thalassemia, who have been in need of new treatments for this lifelong disease,” said Habib Dable, President and Chief Executive Officer of Acceleron. “We are enormously grateful to the patients, families and caregivers who participated in and supported our research. Their contributions have been essential in helping to ensure that Reblozyl would emerge successfully from our longstanding collaboration with Celgene.”
Beta thalassemia is a rare, inherited blood disorder caused by a genetic defect in hemoglobin. The disease is associated with ineffective erythropoiesis, which results in the production of fewer and less healthy RBCs, often leading to severe anemia – a condition that can be debilitating and can lead to more severe complications for patients – as well as other serious health issues. Treatment options for anemia associated with beta thalassemia are limited, consisting mainly of RBC transfusions, which have the potential to contribute to iron overload, which can cause serious complications such as organ damage.
The approval of Reblozyl for beta thalassemia, which received a Priority Review designation from the FDA, is based on results from the pivotal, Phase 3, randomized, double-blind, placebo-controlled, multicenter BELIEVE trial evaluating the safety and efficacy of Reblozyl for the treatment of anemia in adult patients with beta thalassemia who require regular RBC transfusions (defined as 6-20 RBC units per 24 weeks, with no transfusion-free period greater than 35 days during that period). All patients were eligible to receive best supportive care, which included RBC transfusions; iron-chelating agents; use of antibiotic, antiviral, and antifungal therapy; and/or nutritional support, as needed. The trial achieved a clinically meaningful and statistically significant improvement in the primary endpoint. In the Reblozyl arm, 21.4% of patients (n=48) achieved a ≥33% reduction from baseline in RBC transfusion burden (with a reduction of at least 2 units) during weeks 13–24 after randomization, compared to 4.5% (n=5) in the placebo arm (risk difference [95% CI]: 17.0 [10.4, 23.6], P<0.0001).
The study also met key secondary endpoints, including transfusion burden reduction of at least 33% (with a reduction of at least 2 units), during weeks 37 to week 48, which was achieved in 19.6% (n=44) of patients in the Reblozyl arm and 3.6% (n=4) in the placebo arm (risk difference [95% CI]: 16.1 [9.8, 22.4], P<0.0001).
Other efficacy endpoints included transfusion burden reduction of ≥50% (with a reduction of at least 2 units) during weeks 13-24 and weeks 37-48.1 A ≥50% reduction in transfusion burden was observed in 7.6% of patients (n=17) receiving Reblozyl vs. 1.8% of patients (n=2) in the placebo arm at weeks 13-24 (risk difference [95% CI]: 5.8 [1.6, 10.1], P=0.0303), and 10.3% of patients (n=23) vs. 0.9% of patients (n=1) at weeks 37-48 (risk difference [95% CI]: 9.4 [5, 13.7], P=0.0017), respectively.
In the BELIEVE trial, thromboembolic events, including deep vein thromboses, pulmonary embolus, portal vein thrombosis, and ischemic stroke, were experienced in 3.6% (8/223) of Reblozyl treated patients.1 Hypertension was reported in 10.7% (61/571) of Reblozyl-treated patients across the clinical development program.1 Reblozyl may cause fetal harm when administered to a pregnant woman. Serious adverse reactions occurred in 3.6% of patients receiving Reblozyl.1 Serious adverse reactions reported in 1% of patients were cerebrovascular accident and deep vein thrombosis.1 One patient died due to an unconfirmed case of AML.1 The most common adverse reactions (at least 10% for Reblozyl, and 1% more than placebo) were headache (26% vs 24%), bone pain (20% vs 8%), arthralgia (19% vs 12%), fatigue (14% vs 13%), cough (14% vs 11%), abdominal pain (14% vs 12%), diarrhea (12% vs 10%) and dizziness (11% vs 5%).
Permanent discontinuation due to an adverse reaction (Grades 1-4) occurred in 5.4% of patients who received Reblozyl.1 The most frequent adverse reactions requiring permanent discontinuation in patients who received Reblozyl included arthralgia (1%), back pain (1%), bone pain (<1%), and headache (<1%).1 Dosage reductions due to an adverse reaction occurred in 2.7% of patients who received Reblozyl.1 The most frequent adverse reactions requiring a dosage reduction in >0.5% of patients who received Reblozyl included hypertension and headache.1 Dosage interruptions due to an adverse reaction occurred in 15.2% of patients who received Reblozyl.1 The most frequent adverse reactions requiring a dosage interruption in >1% of patients who received Reblozyl included upper respiratory tract infection, ALT increase, and cough.
https://en.wikipedia.org/wiki/Luspatercept