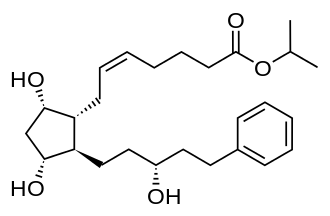
In continuation of my update on Latanoprost
Thea Pharma, Inc. (“Thea”), the U.S. subsidiary of Europe’s leading independent pharmaceutical company, Laboratoires Théa, dedicated to the research, development and commercialization of ophthalmic products, announced U.S. Food and Drug Administration (FDA) approval of the New Drug Application (NDA) of Iyuzeh™ (latanoprost ophthalmic solution) 0.005% for the reduction of elevated intraocular pressure (IOP) in patients with open-angle glaucoma (OAG) or ocular hypertension (OHT).
Iyuzeh™ is the first and only clinically proven formulation of latanoprost available in the United States that is preservative-free. Iyuzeh™ is formulated without any of the preservatives commonly used in topical ocular preparations, including benzalkonium chloride (BAK). Iyuzeh™ has demonstrated consistent IOP-lowering effects and proven tolerability across multiple trials in the U.S. and Europe. In randomized, controlled clinical trials of patients with OAG or OHT with mean baseline IOP of 19-24 mmHg, Iyuzeh™ lowered IOP by 3 – 8 mmHg versus 4 – 8 mmHg by XALATAN® (latanoprost ophthalmic solution) 0.005%, which is preserved with BAK.
“The approval of Iyuzeh™ is a significant milestone for Théa Group, as this is our first FDA approval for a prescription ophthalmic medicine, for our U.S. subsidiary,” said Jean-Frédéric Chibret, President of the Théa Group. “Marketed outside of the U.S. as Monoprost®, the market leading prostaglandin analogue (PGA) in volume, is available in over 46 countries around the world, including France, Germany, Spain, United Kingdom, Italy, and Canada. We are extremely proud to bring our unique preservative-free latanoprost eye drop, Iyuzeh™ to the U.S.
“Our novel patent protected formulation has been made possible by Théa’s innovative scientists. They were able to solve the challenges of solubilizing and stabilizing latanoprost such that Iyuzeh does not need to be manufactured, distributed, or stored at refrigerated temperatures unlike some other competitive brand and generic latanoprost and PGA products. Additionally, Théa is responding to an important unmet need across all stakeholders in the treatment of OAG and OHT. Many patients on preserved glaucoma medications experience moderate to severe signs and symptoms of ocular surface disease (OSD) that can cause discomfort for patients, frustration for physicians, and drive additional costs for payers,” said Susan Benton, Thea’s U.S. President. “We look forward to introducing Iyuzeh™ to U.S. eyecare practitioners in the second half of 2023.”
About Iyuzeh™
Iyuzeh™ (latanoprost ophthalmic solution) 0.005%, an opalescent, white to slightly yellow ophthalmic solution, is a topical formulation of latanoprost that is indicated for the reduction of elevated intraocular pressure (IOP) in patients with open-angle glaucoma (OAG) or ocular hypertension (OHT). Iyuzeh™ does not contain a preservative – it is the first and only preservative-free formulation of latanoprost, the most prescribed prostaglandin F2α analogue (PGA) in the United States. The recommended dosage of Iyuzeh™ is one drop in the affected eye(s) once daily in the evening. If one dose is missed, treatment should continue with the next dose as normal. Reduction of the IOP starts approximately 3 to 4 hours after administration and the maximum effect is reached after 8 to 12 hours. IOP reduction is present for at least 24 hours.
In the two clinical trials conducted with Iyuzeh™ (latanoprost ophthalmic solution) 0.005%, the most frequently reported ocular adverse reactions were conjunctival hyperemia (34%) and eye irritation (19%) compared to XALATAN, the preserved 0.005% latanoprost reference product which reported conjunctival hyperemia (37%) and eye irritation (31%).