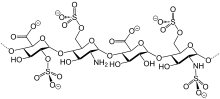
In continuation of my update on Dalteparin
U.S. Food and Drug Administration, approved Fragmin (dalteparin sodium) injection, for subcutaneous use, to reduce the recurrence of symptomatic venous thromboembolism (VTE) in pediatric patients one month of age and older. VTE can include deep vein thrombosis (blood clot in the deep veins of the leg) and pulmonary embolism (blood clot in the lungs), which can lead to death.
“Most children who have VTE are fighting a serious underlying primary illness such as cancer or congenital heart disease. Not only are they fighting a serious illness, having a condition like VTE can then lead to significant complications and even death,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Prior to this approval, there had been no FDA-approved therapies to treat VTE in pediatric patients. Given the unmet need, we granted the Fragmin application priority review and today we are approving it as the first anticoagulant (blood thinner) indicated for pediatric patients. We remain committed to advancing treatments for children with unmet medical needs.”
VTE usually develops as a secondary complication of underlying clinical conditions such as a venous catheter, cancer, infection, congenital heart disease, and trauma or surgery. Pediatric VTE is associated with an increased risk of in-hospital mortality, recurrent VTE and post-thrombotic syndrome (damage to vein).
Fragmin was initially approved by the FDA in 1994 for adults and is a type of heparin, which works as an anticoagulant. The efficacy of Fragmin in children was based on a single trial with 38 pediatric patientswith symptomatic deep vein thrombosis and/or pulmonary embolism. Patients were treated with Fragmin for up to three months, with starting doses by age and weight. At study completion, 21 patients achieved resolution of the qualifying VTE, seven patients showed regression, two patients showed no change, no patients experienced progression of the VTE and one patient experienced recurrence of VTE.
Common side effects of patients taking Fragmin are bleeding, including hemorrhage (heavy discharge of blood from a blood vessel), thrombocytopenia (low blood platelet count), hematoma (collection of blood) or pain at the injection site and transient elevation of transaminases (elevated level of liver enzymes).
Health care professionals are advised to use caution in conditions with increased risk of hemorrhage and monitor thrombocytopenia of any degree closely. Health care professionals are warned not to use benzyl alcohol preservative multiple-dose formulations in infants as they contain benzyl alcohol and should not be used. Patients are advised to have blood count laboratory tests periodically. Health care professionals are advised to monitor patients closely for bleeding when administering Fragmin to patients who currently take anticoagulants. Patients at risk for VTE may receive certain treatments or interventions to help reduce the likelihood of the formation of blood clots (known as thromboprophylaxis), including taking anticoagulants.
The label for Fragmin contains a boxed warning to alert health care professionals and patients that epidural or spinal hematomas (accumulation of blood that can mechanically compress the spinal cord) may occur in patients who are anticoagulated due to taking certain medications called low molecular weight heparins (LMWH) or heparinoids and are receiving neuraxial anesthesia (injection near the spine) or undergoing spinal puncture (removing spinal fluid for testing). These hematomas may result in long-term or permanent paralysis. Health care professionals are advised to consider these risks when scheduling patients for spinal procedures as patients may be at a higher risk of developing VTE. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include: use of indwelling epidural catheters, use of other drugs that affect hemostasis at the same time when using Fragmin, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors and other anticoagulants; history of traumatic or repeated epidural or spinal punctures; and a history of spinal deformity or surgery. The optimal timing between the administration of Fragmin and neuraxial procedures is not known. Health care professionals are advised to monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary. Health care professionals are advised to consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis.
https://en.wikipedia.org/wiki/Dalteparin_sodium
https://www.drugbank.ca/drugs/DB06779