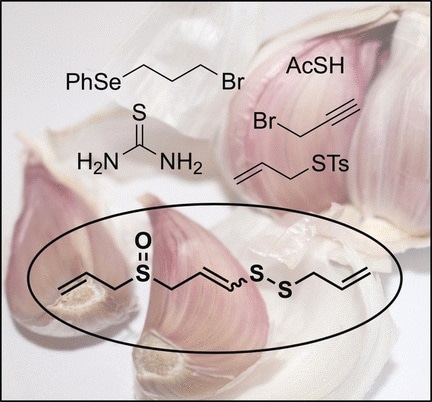
Fresh garlic extracts contain a variety of healthy organosulfur compounds, among which ajoene forms a major oil-extractable ingredient. Now, chemists in the United Kingdom have synthesized ajoene from readily available components for the first time. The results, which are published in the journal Angewandte Chemie, show that ajoene is accessible on a large scale with very few synthetic steps. Chemical synthesis of biologically active compounds is important for their further evaluation in medicinal research.
If garlic is cut or chewed, enzymes present in the damaged tissue start to degrade its main organosulfur metabolite, alliin. The first degradation product is allicin, which gives fresh garlic preparations their characteristic pungent odor. However, this molecule decomposes further into various, largely oil-soluble compounds, all characterized chemically as organosulfides or disulfides. A more stable decomposition product and main component in oil extracts is ajoene. This compound has similar health-promoting effects to allicin and it exhibits anticancer activity.
Although ajoene can be isolated from garlic extracts, chemical synthesis would have many advantages. Synthesized ajoene would allow the introduction of chemical modifications, a key provision in drug research. Therefore, Thomas Wirth and his group at Cardiff University in collaboration with the Welsh company Neem Biotech in the United Kingdom have now developed a fully synthetic approach based on simple, readily available components. The sequence starts with a simple dibromide and terminates with the oxidation of an organoselenium compound. Oxidative elimination of the selenium compound, the scientists noted, leads to the formation of the terminal carbon-carbon double bond characteristic for the ajoene molecule. At the same time, its sulfide moiety is oxidized to a sulfoxide, another characteristic chemical function in ajoene.
The biggest challenge in ajoene synthesis was minimizing the various side reactions typical for organosulfur compounds, Wirth and his team reported. Such side reactions profoundly decreased the yield in the biomimetic approach to ajoene, which started from allicin. But low yields turned out to be a problem in total synthesis as well. Therefore, the scientists explored several modifications in the reaction steps, but the most profound improvement, unexpectedly, came from scaling up the synthesis. On the 200-gram scale, the final oxidation yielded 56 percent of the product, the authors reported, which was twice as much as when working on the milligram scale.
The product was biologically active. Testing its activity against bacteria in a bioassay, Wirth and his group found that synthetic ajoene performed similarly to or even better than natural ajoene extracted from garlic. It inhibited biological communication called quorum sensing in Gram-negative bacteria, which may lead to biofilm formation. Inhibiting this could be a promising usage of ajoene, the authors suggested. And as total synthesis has now made this compound more easily accessible, its career in medicinal chemistry may be ready to take off.
Ref : https://newsroom.wiley.com/press-release/angewandte-chemie-international-edition/garlic-ingredient-lab-bench-short-total-synthe