In continuation of my update on Fycompa
Eisai Inc. announced that the U.S. Food and Drug Administration (FDA) has approved Fycompa (perampanel) CIII Oral Suspension as adjunctive therapy for the treatment of partial-onset seizures (POS) with or without secondarily generalized seizures, and primary generalized tonic-clonic (PGTC) seizures in patients with epilepsy 12 years of age and older. The oral suspension formulation is a bioequivalent, interchangeable alternative to the Fycompa tablet formulation, and is expected to be available to patients in June 2016.
"We are excited about the approval of Fycompa Oral Suspension, as it gives another option to patients with epilepsy who may have difficulty swallowing tablets or prefer liquids," said Lynn Kramer, M.D., Chief Clinical Officer and Chief Medical Officer, Neurology Business Group, Eisai. "The development of this new formulation underscores Eisai's commitment to advancing epilepsy care by making contributions to help address the diversified needs of epilepsy patients and their families."
The approval of Fycompa Oral Suspension was based on a bioequivalence (BE) study that demonstrated BE between a single dose of perampanel oral suspension and a single dose of perampanel tablet, when administered under fasted conditions in healthy subjects.
About Fycompa
Fycompa (perampanel) is indicated as adjunctive therapy for the treatment of partial-onset seizures with or without secondarily generalized seizures and primary generalized tonic-clonic seizures in patients with epilepsy 12 years of age and older.
Fycompa is an oral medication and the first and only FDA-approved non-competitive AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor antagonist. The precise mechanism by which Fycompa exerts its antiepileptic effects in humans has not been fully elucidated.
Fycompa is supplied as 2 mg, 4 mg, 6 mg, 8 mg, 10 mg and 12 mg film-coated tablets, and now in an oral suspension formulation. Fycompa has been designated by the U.S. Drug Enforcement Administration as a federally-controlled substance (CIII).
Fycompa, approved in 43 countries, was discovered and developed by Eisai. Over 60,000 patients globally have been treated with Fycompa.




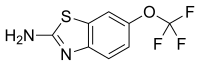 riluzole
riluzole
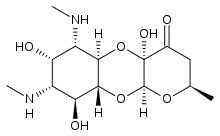
 Apramycin
Apramycin Vernakalant
Vernakalant  Ibutilide
Ibutilide