“We are thrilled with the FDA's approval of Recorlev as a safe and effective treatment option for patients with endogenous Cushing's syndrome. With this approval, Xeris’ experienced endocrinology-focused commercial organization can begin rapidly working to help address the needs of Cushing’s syndrome patients in the U.S. who are treated with prescription therapy,” said Paul R. Edick, Chairman and CEO of Xeris Biopharma. “Today’s announcement also reinforces the value that we saw in acquiring Strongbridge Biopharma’s attractive rare disease portfolio, which we believe will deliver compelling long-term value to our shareholders. We look forward to making Recorlev commercially available in the first quarter.”
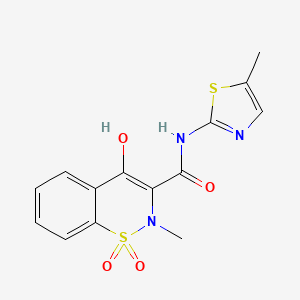
The approval of Recorlev was based upon safety and efficacy data from two positive Phase 3 studies that evaluated a combined study population of 166 patients, which was representative of the adult drug-treated U.S. population with Cushing’s syndrome. The SONICS study met its primary and key secondary endpoints, significantly reducing and normalizing mean urinary free cortisol concentrations without a dose increase. LOGICS, a double-blind, placebo-controlled randomized-withdrawal study that met its primary and key secondary endpoints, confirmed the efficacy and safety of Recorlev in normalizing and maintaining therapeutic response compared with placebo.
“Levoketoconazole (Recorlev) is an important and welcome new therapeutic option for clinicians to help manage patients with endogenous Cushing's syndrome, a severe, potentially life-threatening rare disease, if not appropriately treated, with multisystem signs and symptoms,” said Maria Fleseriu, M.D., FACE, professor of Medicine and Neurological Surgery and director of the Pituitary Center at Oregon Health Sciences University. “In prospective clinical studies, treatment with levoketoconazole was shown to be effective for reducing and normalizing cortisol.”
“Cushing’s syndrome is a rare disease that can be physically and emotionally devastating to the patient. Most patients endure years of symptoms prior to obtaining a diagnosis and are then faced with limited effective treatment options," said Leslie Edwin, president of the Cushing’s Support & Research Foundation. “Today we are excited to see that the long and complicated path of rare drug development has reached FDA approval on a new therapeutic option for our underserved Cushing's community. We are grateful that the researchers worked so diligently for so long to establish the safety and efficacy of this drug. Rare disease patients know the importance of sharing their complicated experiences as ‘expert witnesses’, and we thank Xeris for being an early adherent to this concept. We especially want to thank the clinical trial patients who made this progress possible.”
Xeris is committed to ensuring everyone who needs access to their therapies will receive it. Xeris has created Xeris CareConnection™ to provide a comprehensive program for patients and their caregivers throughout the treatment journey, including financial assistance, one-on-one support, and educational resources. Xeris CareConnection also supports healthcare professionals and their teams through education on access and reimbursement. To get started with Recorlev, reach out to Xeris CareConnection (available Monday–Friday from 8 a.m–7 p.m ET) at 1-844-444-RCLV (7258).