
In continuation of my update on VRAYLAR (cariprazine)

Allergan plc (NYSE: AGN) and Gedeon Richter Plc. announced that the U.S. Food and Drug Administration (FDA) has approved a supplemental New Drug Application (sNDA) for Vraylar (cariprazine) for expanded use to treat depressive episodes associated with bipolar I disorder (bipolar depression) in adults. Vraylar is also approved in the U.S. to treat manic or mixed episodes associated with bipolar I disorder in adults. There are nearly 11 million adults in the U.S. living with bipolar disorder, a condition that causes extreme shifts in mood, energy, and activity levels.
"Treating bipolar disorder can be very difficult because people living with the illness experience a range of depressive and manic symptoms, sometimes both at the same time, and this FDA approval gives healthcare providers a new option to treat the full spectrum of bipolar I disorder symptoms, specifically manic, mixed, and depressive episodes, with just one medication," said Dr. Stephen Stahl, Professor of Psychiatry at the University of California San Diego and lead author of the post hoc analysis, Cariprazine Efficacy in Patients with Bipolar Depression and Concurrent Manic Symptoms. "Treating depression, mania and mixed episodes with a single medication is important for people living with, and healthcare providers treating, this complex illness. This approval can streamline a treatment decision while helping to stabilize the disorder."
Seventy percent of people living with bipolar disorder receive at least one misdiagnosis and consult an average of four doctors over approximately 10 years before being accurately diagnosed.3 Many patients take multiple medications to treat the symptoms of this condition.
The FDA approval for the expanded indication of Vraylar is based on three pivotal trials, including RGH-MD-53, RGH-MD-54 and RGH-MD-56, in which cariprazine demonstrated greater improvement than placebo for the change from baseline to week six on the Montgomery Asberg Depression Rating scale (MADRS) total score. In all three studies, the Vraylar 1.5 mg dose demonstrated statistical significance over placebo; additionally, in RGH-MD-54, the Vraylar 3 mg dose demonstrated statistical significance over placebo. Common adverse events reported in the pivotal trials were nausea, akathisia, restlessness, and extrapyramidal symptoms.
"This approval represents an important milestone in our efforts to help patients and prescribing healthcare providers effectively manage bipolar I disorder and demonstrates our ongoing focus on mental health," said David Nicholson, Chief Research & Development Officer at Allergan. "We are committed to developing therapies for complex mental health disorders, including Vraylar, which is currently in Phase 3 clinical trials for the treatment of major depressive disorder."
"This approval is considered a notable achievement in the development process of cariprazine, our flagship product," said Dr. István Greiner, Research Director of Gedeon Richter Plc. "We are pleased that more and more patient groups suffering from psychiatric disorders will get access to cariprazine as a treatment option."
About Vraylar (cariprazine)
Vraylar is an oral, once daily atypical antipsychotic approved for the acute treatment of adults with manic or mixed episodes associated with bipolar I disorder (3 to 6 mg/day) and for the treatment of depressive episodes associated with bipolar I disorder (bipolar depression) in adults (1.5 or 3 mg/day). Vraylar is also approved for the treatment of schizophrenia in adults (1.5 to 6 mg/day).
While the mechanism of action of Vraylar is unknown, the efficacy of Vraylar could be mediated through a combination of partial agonist activity at central dopamine D₂ and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors. Pharmacodynamic studies with cariprazine have shown that it acts as a partial agonist with high binding affinity at dopamine D3, dopamine D2, and serotonin 5-HT1A receptors. Cariprazine demonstrated up to ~8-fold greater in vitro affinity for dopamine D3 vs D2 receptors. Cariprazine also acts as an antagonist at serotonin 5-HT2B and 5-HT2A receptors with high and moderate binding affinity, respectively as well as it binds to the histamine H1 receptors.
Vraylar shows lower binding affinity to the serotonin 5-HT2C and α1A- adrenergic receptors and has no appreciable affinity for cholinergic muscarinic receptors. The clinical significance of these in vitro data is unknown.
Vraylar was discovered and co-developed by Gedeon Richter Plc and is licensed by Allergan, in the U.S. and Canada. For more than a decade both companies have conducted over 20 clinical trials enrolling thousands of patients worldwide to evaluate the efficacy and safety of cariprazine for people living with a broad range of mental health illnesses.


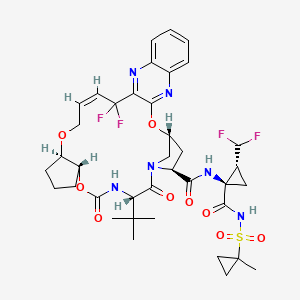
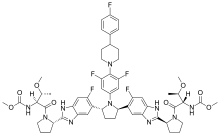 Pibrentasvir
Pibrentasvir
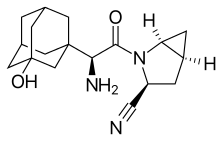 Saxagliptin and
Saxagliptin and  (Metformin)
(Metformin)


