After treatment with riluzole, the brains of old rats showed more of a transporter molecule that removes excess glutamate, (green fluorescence, right) as compared to untreated rats (left).
Aging takes its toll on the brain, and the cells of the hippocampus--a brain region with circuitry crucial to learning and memory--are particularly vulnerable to changes that can lead to Alzheimer's disease or cognitive decline. With the hope of counteracting the changes that can lead to these two conditions, researchers at Rockefeller University and their colleagues have begun examining the effects of a drug known to affect this circuitry.
In new research described recently in Molecular Psychiatry, a team led by Ana Pereira, Instructor in Clinical Medicine in Bruce McEwen's laboratory found that the drug, riluzole, is capable of reversing key genetic changes associated with these conditions.
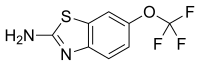 riluzole
riluzole
"In aging and Alzheimer's, the chemical signal glutamate can accumulate between neurons, damaging the circuitry," Pereira says. "When we treated rats with riluzole, we saw a suite of changes. Perhaps most significantly, expression of molecules responsible for clearing excess glutamate returned to more youthful levels."
Previous work in McEwen's lab by Pereira has shown that the drug prompted structural changes in rats' neurons that prevent the memory loss often seen in old animals. Pereira is currently testing riluzole for the first time in Alzheimer's patients in a clinical trial at the Rockefeller University Hospital.
Glutamate clean up
Generally, glutamate is released to excite other neurons and doesn't linger in the spaces between them. As we age, though, the system gets a little leaky and glutamate can build up in these intercellular spaces. This happens in part when neurons make less and less of the transporter molecule responsible for removing excess glutamate. When it accumulates, this essential neurotransmitter can cause big problems, damaging or killing neurons and so contributing to Alzheimer's disease, and other disorders.
Pereira and co-first author Jason Gray, a postdoc in the lab sought to better understand the molecular vulnerabilities of an aging glutamate system and riluzole's effect on it.
"The essence is we used a drug known to modulate glutamate, and when we gave it to old rats, we saw it reversed many of the changes that begin in middle age in the hippocampus," Gray says. "We saw a similar pattern when we compared the riluzole-induced changes to data from Alzheimer's patients--in a number of key pathways in the hippocampus, the drug produced an effect opposing that of the disease."
The drug, it turns out, modifies the activity of certain genes in an aged animal to resemble that of a younger rat. For example, the researchers found that the expression of a gene called EAAT2, which has been linked to Alzheimer's and is known to play a role in removing excess glutamate from nerve fibers, declines as the animals age. However, in rats treated with riluzole this gene's activity was brought back to its youthful levels.
New targets for treatments?
In addition to its potential ability to allay memory loss and cognitive decline, riluzole is attractive as a potential treatment for Alzheimer's. The drug is already being used to treat another neurological disease, amyotrophic lateral sclerosis, and is therefore considered relatively safe. In Pereira's ongoing clinical trial, patients with Alzheimer's disease have thus far been treated with either the drug or a placebo, and have been undergoing tests to help determine whether their brain functions have been improved.
"We hope to use a medication to break the cycle of toxicity by which glutamate can damage the neurons that use it as a neurotransmitter, and our studies so far suggest that riluzole may be able to accomplish this," Pereira says. "We found that in addition to recovering the expression of EAAT2, the drug restored genes critical for neural communication and plasticity, both of which decline with aging and even more significantly in Alzheimer's disease."
The findings also help to lay the groundwork for further study of glutamate transporters as potential targets for treating both conditions.
Ref : http://newswire.rockefeller.edu/2016/05/02/an-experimental-alzheimers-drug-reverses-genetic-changes-thought-to-spur-the-disease/
Ref : http://www.nature.com/mp/journal/vaop/ncurrent/full/mp201633a.html

 Apramycin
Apramycin Vernakalant
Vernakalant  Ibutilide
Ibutilide