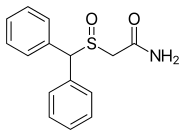
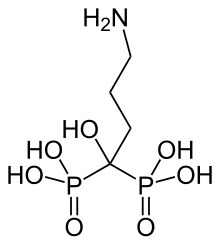
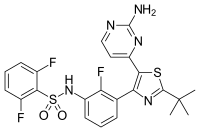
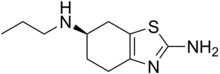
In continuation of my update on ebselen
An international study led by biochemists at the University of Liverpool has shown that the drug-molecule ebselen can correct many of the toxic characteristics of a protein that causes some cases of hereditary motor neurone disease (MND).
MND is an incurable, progressive disease that attacks the nerves controlling movement so muscles no longer work. MND affects about 5000 people in the UK at any one time and present treatment options have only a modest effect in improving the patient's quality of life.

Inherited MND is a rare form of the disease (5-10% of total cases) that runs in families. Around 20% of hereditary MND cases are caused by mutations in a gene which codes for a protein called SOD1. When the SOD1 gene is mutated, the protein assembly process malfunctions and steps are missed out. This makes the SOD1 protein structurally unstable leading to formation of protein 'clumps' in the motor neurones, causing them to die.
In a paper published in Nature Communications, scientists from the Universities of Liverpool (UK), Florence (Italy) and Wollongong (Australia) used state-of-the-art crystallography, mass-spectrometry and in-cell NMR technologies to search for a drug molecule which could 'correct' the SOD1 assembly line.
They found that ebselen, a drug which was discovered in the 1980s and has been investigated as a potential treatment for a variety of nervous system disorders, can effectively restore several important steps in the SOD1 assembly process including folding, dimerization and zinc binding.
Dr. Gareth Wright, an MND researcher at the University of Liverpool, said: "This discovery has the potential to prevent the accumulation of SOD1 into the large aggregates we see within the motor neurons of effected individuals. If we can stop that, we might be able to stop the neurons dying."
Professor Samar Hasnain, a structural biologist at the University of Liverpool, added: "The next step is to test ebselen in settings more accurately resembling human neuronal cells and optimising it so that it can become useful as a drug for motor neuron disease."
Commenting on the study, Dr Brian Dickie, Director of Research Development at the Motor Neurone Disease Association, said: "A causal link between the SOD1 gene and certain forms of hereditary motor neuron disease was established a quarter of a century ago. It is very encouraging to see new therapeutic strategies starting to emerge from the considerable advances in scientific understanding that have occurred in recent years."