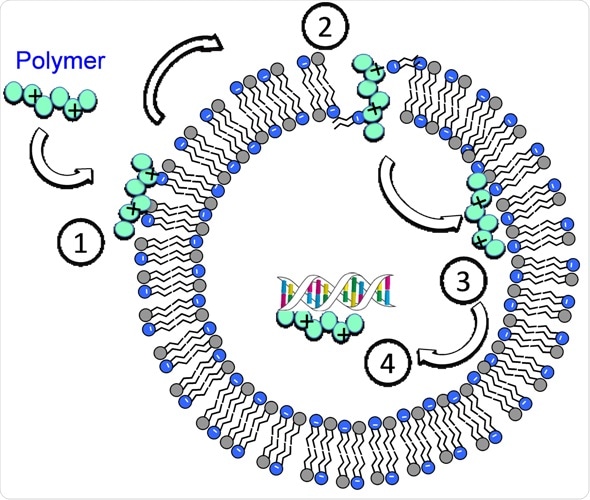
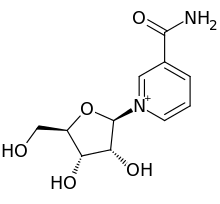
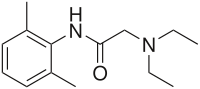
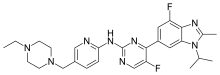
In continuation of my update on vancomycin

CutisPharma announced the US Food and Drug Administration (FDA) approval of Firvanq (vancomycin hydrochloride) for oral solution, for the treatment of Clostridium difficile associated diarrhea and enterocolitis caused by Staphylococcus aureus, including methicillin-resistant strains.
“We are pleased to announce the FDA approval of Firvanq,” said Neal I. Muni, MD, MSPH, Chief Executive Officer of CutisPharma. “Firvanq's approval is an important step forward to providing patients the only FDA-approved vancomycin oral liquid treatment option for Clostridium difficile associated diarrhea, a life-threatening condition that affects over a half-million patients in the United States annually.”
Upon its launch, which is targeted to be April 2, 2018, Firvanq will replace CutisPharma’s FIRST®-Vancomycin Unit-of-Use Compounding Kit, which has been available to pharmacists that need a convenient, accurate, and compliant way to compound vancomycin oral liquid therapy. Firvanq will be commercially available in 25 mg/mL and 50 mg/mL strengths in convenient 150 mL and 300 mL sizes. Firvanq is designed to be easy to use and has the potential to be a cost-effective alternative to existing vancomycin therapies.
“As a practicing infectious disease physician treating many patients with CDAD, having an FDA-approved vancomycin oral liquid formulation that is affordable and accessible to my patients is very beneficial,” said Stuart Johnson, MD, Loyola University Medical Center. “Patient access is currently limited by the fact that only a select few pharmacies perform compounding in the outpatient setting these days, given the many new regulations in place. Availability of an FDA-approved vancomycin oral liquid treatment will effectively allow any pharmacy to stock this therapy, and hopefully encourage third-party payer reimbursement, significantly improving accessibility and convenience for patients.”