“An effective antiretroviral regimen can be devised for most people living with the virus; however, some people living with HIV no longer have durable viral suppression due to resistance to multiple classes of antiretroviral therapies,” said Sorana Segal-Maurer, MD, Director of the Dr. James J. Rahal Jr. Division of Infectious Diseases at NewYork-Presbyterian Queens, Professor of Clinical Medicine at Weill Cornell Medicine and the Site Principal Investigator for the CAPELLA trial. “The availability of new classes of antiretroviral drugs is critical for heavily treatment-experienced people with multi-drug resistant HIV. Following today’s decision from the FDA, lenacapavir helps to fill a critical unmet need for people with complex prior treatment histories and offers physicians a long-awaited twice-yearly option for these patients who otherwise have limited therapy choices."
Despite the significant advances in ARV therapy, there remain numerous critical and pressing unmet needs for people living with HIV. This is particularly true for individuals who are heavily treatment experienced – which account for an estimated 2% of adults living with HIV who are on treatment globally –- and are unable to maintain virologic suppression due to resistance, intolerance or safety considerations. This type of complexity further increases the chance of treatment failure, underscoring the need for new treatment options that are active against resistant variants of the virus with a novel mechanism of action.
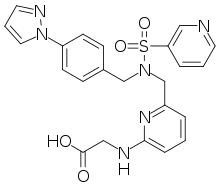
Lenacapavir is a breakthrough innovation with the potential to be a preferred and versatile foundational long-acting agent due to its therapeutic potency and a range of dosing frequencies and routes of administration. Lenacapavir is being developed as a foundation for Gilead’s future HIV therapies with the goal of offering several long-acting options that help address individual needs and preferences that may help optimize outcomes and reduce burden of care. Lenacapavir is being studied in multiple ongoing early and late-stage development programs and has the potential to offer a diverse set of person-centric options for treatment and prevention that could uniquely fit into the lives of people living with HIV and people who would benefit from pre-exposure prophylaxis (PrEP). The use of lenacapavir for HIV prevention is investigational and the safety and efficacy of lenacapavir for this use have not been established.
“This news is an important milestone in the work to help end the HIV epidemic as Sunlenca is now the only FDA-approved twice-yearly treatment for people with multi-drug resistant HIV,” said Daniel O’Day, Chairman and Chief Executive Officer, Gilead Sciences. “Gilead scientists have developed a unique and potent antiretroviral medicine in Sunlenca with the potential for flexible dosing options. Our goal is to deliver multiple long-acting options for treatment and prevention that are tailored to the needs of people living with HIV and people who could benefit from PrEP medicines.”
The FDA approval for Sunlenca is supported by data from the Phase 2/3 CAPELLA trial, which evaluated lenacapavir in combination with an optimized background regimen in people with multi-drug resistant HIV-1 who are heavily treatment experienced. CAPELLA participants had undergone previous treatment with a median of nine antiretroviral medications. In this patient population with significant unmet medical need, 83% (n=30/36) of participants randomly allocated to receive lenacapavir in addition to an optimized background regimen achieved an undetectable viral load (<50 copies/mL) at Week 52. Additionally, these participants achieved a mean increase in CD4 count of 82 cells/µL. These data were presented at the 29th Conference on Retroviruses and Opportunistic Infections (virtual CROI 2022).
Sunlenca was reviewed and approved as a medication with FDA Breakthrough Therapy Designation, which is intended to expedite the development and review of new drugs which may demonstrate substantial improvement over available therapy. In May 2019, the FDA granted Breakthrough Therapy Designation for the development of lenacapavir for the treatment of HIV-1 infection in heavily treatment-experienced patients with multi-drug resistance in combination with other antiretroviral drugs.
Lenacapavir, alone or in combination, is not approved by any regulatory authority outside of the United States, United Kingdom, Canada or the European Union for any use. The European Marketing Authorization applies to all 27 member states of the European Union, as well as Norway, Iceland and Liechtenstein.
The use of lenacapavir for HIV prevention is investigational and the safety and efficacy of lenacapavir for this use have not been established. Gilead is studying the safety and efficacy of lenacapavir for HIV prevention in multiple ongoing clinical studies.
Ref : https://en.wikipedia.org/wiki/Lenacapavir