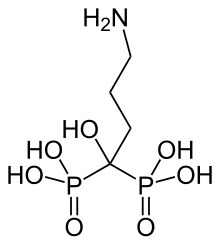
In continuation of my update on Rucaparib
A targeted therapy that has shown its power in fighting ovarian cancer in women including those with BRCA1 and BRCA2 mutations may also help patients with aggressive pancreatic cancer who harbor these mutations and have few or no other treatment options. An international team of researchers led by the Perelman School of Medicine and the Basser Center for BRCA at the University of Pennsylvania reported their findings this week in JCO Precision Oncology.
The drug, PARP inhibitor rucaparib, which was approved by the U.S. Food and Drug Administration (FDA) last month for the treatment of women with ovarian cancer who have recurrent disease or received prior therapies, showed its clinical benefit in previously treated pancreatic patients with BRCA mutations in a phase II clinical trial. Of the 19 patients treated, four had responses and two additional patients had stable disease.
"These results not only point us in a new treatment direction to further investigate for patients with pancreatic cancers, but it also reinforces the clinical significance of the BRCA genes beyond ovarian and breast cancer and the utility of PARP inhibitors in other cancers," said Susan M. Domchek, MD, executive director of the Basser Center for BRCA at the Abramson Cancer Center of the University of Pennsylvania.
PARP—poly (ADP-ribose) polymerase—is an enzyme used by healthy cells to repair themselves. However, cancer cells also use PARP to repair DNA damage, thus extending their growth and possible lethality. Preliminary results from the study, which included patients from seven centers around the globe, were presented at the annual meeting of the American Society of Clinical Oncology in 2016. These latest findings represent the full study.
Pancreatic cancer, which is often caught in later, more aggressive stages, is projected to become the second leading cause of cancer death by 2020, emphasizing the need for a larger and more effective arsenal of treatments to combat the disease. Only about 32 percent of patients respond to a first line of chemotherapy, and less than 20 percent who don't respond to a first line of chemotherapy end up responding to a second.
This underscores the importance of looking outside of chemotherapy options, the authors said, particularly in patients with targetable mutations, like BRCA.
Importantly, Domchek said, none of the patients who benefited from rucaparib had tumors that had progressed on a prior platinum-based chemotherapy, suggesting a potential role for rucaparib as an earlier treatment for patients whose tumors are not resistant to such treatments.
Rucaparib is a PARP inhibitor shown to be an effective therapy in ovarian cancers with BRCA 1/2 mutations. In 2016, the drug was approved by the FDA for women with BRCA-associated ovarian cancer who received two or more prior chemotherapies. And in April 2018, the approval was extended to women with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are having a complete or partial response to platinum-based chemotherapies.
The success of rucaparib in ovarian cancers is what prompted the clinical study in pancreatic patients with the same mutation. About nine percent of pancreatic patients have BRCA/2 mutation associated pancreatic cancer.
Overall, a clinical benefit was observed in 32 percent of patients (6/19) treated with rucaparib, and 45 percent in patients (4/9) who had received only one prior chemotherapy for locally advanced or metastatic disease. Nine patients had progressive disease, and three were not evaluable for response. The objective confirmed response rate, the primary endpoint for the study, was 16 percent (3/19).
The trial included 11 men and eight women, with a median age of 57. Twenty-one percent of the patients had BRCA1mutation-associated pancreatic cancer, while 79 percent were associated with BRCA2 mutations.
"Consideration should be given to use of this therapy for treatment of patients whose tumors have not progressed on prior platinum therapy," the authors wrote. "Future studies should focus on better understand the sequencing of PARP inhibitor treatment and potential maintenance therapy, as well as potential predictors of resistance to therapy."