
BeyondSpring Pharmaceuticals announced the receipt of a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) for the New Drug Application (NDA) seeking approval of plinabulin in combination with granulocyte colony-stimulating factor (G-CSF) for the prevention of chemotherapy-induced neutropenia (CIN). The FDA issued the CRL to indicate that they have completed their review of the application and have determined that it cannot be approved in its present form.

The FDA’s CRL indicated that the results of the single registrational trial (106 Phase 3) was not sufficiently robust to demonstrate benefit and that a second well controlled trial would be required to satisfy the substantial evidence requirement to support the CIN indication.
“BeyondSpring strongly believes that plinabulin in combination with G-CSF has significant potential to raise the standard of care in CIN, a devastating side effect of chemotherapy,” said Dr. Lan Huang, BeyondSpring’s co-founder, chief executive officer and chairwoman. “The Company plans to request a meeting with the FDA and remains committed to its goal of bringing plinabulin to cancer patients in need globally.”
BeyondSpring remains confident in the efficacy and safety data for plinabulin in combination with G-CSF for the prevention of CIN. The Company expects to work closely with the FDA to consider the possible future clinical pathway for CIN, which may include a second study.
Plinabulin is the first drug candidate submitted for FDA approval that has the potential to work in the critical first week of chemotherapy treatment before G-CSF is effective, to prevent the onset and improve clinical outcomes of CIN.
Plinabulin, BeyondSpring’s lead asset, is a selective immunomodulating microtubule-binding agent (SIMBA), which is a potent antigen presenting cell (APC) inducer. It is a novel, intravenous infused, patent-protected, NDA-stage asset for CIN prevention and a Phase 3 anti-cancer candidate for non-small cell lung cancer (NSCLC) with recently released positive topline data. Plinabulin triggers the release of the immune defense protein, GEF-H1, which leads to two distinct effects: first is a durable anticancer benefit due to the maturation of dendritic cells resulting in the activation of tumor antigen-specific T-cells to target cancer cells, and the second is early-onset of action in CIN prevention after chemotherapy by boosting the number of hematopoietic stem/progenitor cells (HSPCs). Plinabulin received Breakthrough Therapy designation and priority review from both U.S. and China FDA for the CIN prevention indication. As a “pipeline in a drug,” plinabulin is being broadly studied in combination with various immuno-oncology agents that could boost the effects of the PD-1/PD-L1 antibodies and re-sensitize PD-1/PD-L1 antibody-resistant patients.
https://en.wikipedia.org/wiki/Plinabulin
















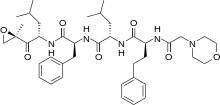 Carfilzomib
Carfilzomib 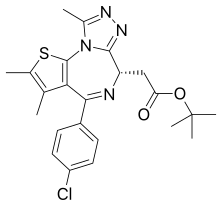 JQ1
JQ1  ABT-199
ABT-199
