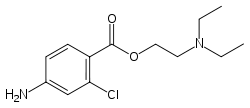
Harrow (Nasdaq: HROW), an eyecare pharmaceutical company exclusively focused on the discovery, development, and commercialization of innovative ophthalmic therapies, and Sintetica, S.A., a growing pharmaceutical company focused on analgesics, local anesthetics, and sterile injectable solutions, announced the U.S. Food and Drug Administration (FDA) approval of Iheezo (chloroprocaine hydrochloride ophthalmic gel) 3% for ocular surface anesthesia. Iheezo is a sterile, single-patient‑use, physician‑administered, ophthalmic gel preparation, containing no preservatives, that is safe and effective for ocular surface anesthesia. Iheezo represents the first approved use in the U.S. ophthalmic market of chloroprocaine hydrochloride and the first branded ocular anesthetic approved for the U.S. ophthalmic market in nearly 14 years. Iheezo is protected by an Orange Book-listed patent that is valid until 2038.
“On behalf of all our ophthalmic physician partners and the patients they serve, we and our partners at Sintetica are grateful to the FDA for a New Drug Application (NDA) review process that resulted in the approval of Iheezo in advance of our PDUFA target action date,” said Mark L. Baum, Harrow Chairman and Chief Executive Officer. “We have always believed in the unique clinical value of Iheezo, and now that Iheezo is approved for use in the U.S. market, it has the potential to become an indispensable premium tool for eyecare professionals and their patients requiring ocular surface anesthesia.”
Nicola Caronzolo, Sintetica Chief Executive Officer, added, “I am particularly proud of this important milestone, which exemplifies the quality of Sintetica’s research and development groups and our ability to innovate – to be a global pharmaceuticals leader. I want to give special thanks to our regulatory group, who while working with the Harrow team, performed extraordinarily well, resulting in this early U.S. market approval for this important new medicine.”
The safety and efficacy of Iheezo were demonstrated in three human clinical studies. Studies 1 and 2 were randomized, double-blinded, placebo-controlled studies that evaluated the effect of Iheezo on healthy volunteers, and Study 3 was a randomized, prospective, multi-center, active-controlled, observer‑masked study that evaluated the administration of Iheezo in patients undergoing cataract surgery. Study 3 marks the first time a U.S. drug candidate was studied in a surgical model for FDA approval in the ocular surface anesthesia category. This study demonstrated that Iheezo not only worked rapidly (about 1 to 1.5 minutes) and provided sufficient anesthesia to successfully perform the surgical procedure (on average lasting 22 minutes), but importantly, no patient dosed with Iheezo required a supplemental treatment to complete the surgical procedure.
According to a September 2021 report by Market Scope, there are an estimated 4.5 million cataract surgeries and over 8 million intravitreal injections performed annually in the U.S., all of which typically utilize some form of ocular surface anesthesia.
Baum continued, “Harrow currently provides perioperative medications for a significant number of the U.S. ophthalmic surgical procedures. We believe our customer base of more than 10,000 ophthalmologists, optometrists, retina specialists, outpatient hospital facilities, and ambulatory surgery centers will appreciate the unique clinical value and practice efficiency Iheezo offers, including its single‑use packaging format, which according to the Institute for Safe Medication Practices (ISMP), decreases the risk of infection and medication errors associated with the use of communal eye drops.
“We have been planning for the commercial launch of Iheezo for over a year, and with our national market access and sales organization already in place, we are 100% ready. Given our earlier FDA approval date, we have accelerated our market access strategy to support a commercial launch date slightly ahead of our previously planned launch at the May 2023 American Society of Cataract and Refractive Surgery (ASCRS) meeting in San Diego, CA.”
https://en.wikipedia.org/wiki/Chloroprocaine