In recent years, hospitals have reported dramatic increases in the number of cases of the highly contagious, difficult-to-treat, and often deadly antibiotic-resistant bacteria carbapenem-resistant Enterobacteriaceae (CRE). Now, investigators at Beth Israel Deaconess Medical Center (BIDMC) have developed a promising method of identifying new antimicrobials that target these organisms. The research is published in April issue of the journal ASSAY and Drug Development Technologies.
CRE are Gram-negative bacteria that frequently express a gene that codes for carbapenemase--an enzyme that breaks down carbapenem and other antibiotics--and that is located on "mobile genetic elements" called plasmids, which can jump from one bacterium to another. The two most common types of CRE are carbapenem-resistant Klebsiella species and carbapenem-resistant Escherichia coli. Patients who become infected with these bacteria have few antibiotic treatment options.
"The US Centers for Disease Control and Prevention recently classified these carbapenem-resistant organisms in their highest, most urgent antimicrobial resistance threat level," said James Kirby, MD, Director of the Clinical Microbiology Laboratory at BIDMC and an Associate Professor of Pathology at Harvard Medical School. "Unfortunately, often either no effective or only toxic antimicrobial options remain for CRE treatment. Moreover, CRE are particularly frightening as they are now increasing in prevalence across the United States and the world."
While there is a critical need for new antimicrobial agents against CRE and other emerging antibiotic-resistant bacteria, the number of new antibiotics that have been developed and approved has steadily decreased in recent decades. To identify new or existing drugs that can destroy multidrug-resistant CRE, Kirby and postdoctoral fellow Kenneth Smith, PhD examined approximately 10,000 compounds with known activity--so called known bioactive molecules--including most previously FDA-approved drugs, veterinary drugs and inhibitors of various cellular processes not currently used as therapeutics.
Through a process called high throughput screening, the investigators looked to see whether any of these compounds could either directly inhibit the growth of CRE or restore the effectiveness of carbapenem against these organisms.
From these screening experiments, 79 compounds were found to inhibit CRE. Of these, three had already been approved for human and veterinary use: azidothymidine (also known as AZT, a therapy for HIV infection), spectinomycin (a treatment for gonorrhea infection) and apramycin (a veterinary antimicrobial). When tested against a large number of CRE strains, the three compounds were broadly active against the strains.
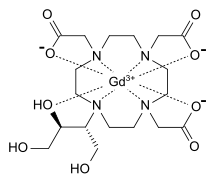
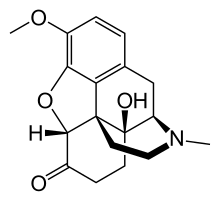
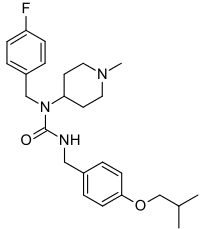
 azidothymidine,
azidothymidine,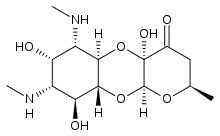 Spectinomycin
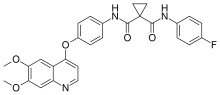
Spectinomycin Apramycin
Apramycin
"These antimicrobials currently have other intended uses and are not currently considered as treatments for CRE, however our findings suggest they could potentially be repurposed for CRE treatment," said Smith. Apramycin and spectinomycin are of particular interest because they have minimal side effects, making them potentially ideal new therapeutic options for CRE infection.
Smith added that while these drugs might be used by themselves to treat CRE infection, they could also be used as starting points for further drug development. "Specifically, these antibiotics could be structurally modified to further increase their activity and prevent resistance from developing against them," he explained.
The next step in this line of research is to examine the potency of the identified drugs in an animal model of CRE infection. "We are also using the same high throughput screening technology to investigate a collection of more than 200,000 completely novel compounds with as yet uncharacterized biological activity in the hopes of identifying new classes of compounds with potent activity against CRE and other multi-drug resistant pathogens," Kirby said.
Ref : http://online.liebertpub.com/doi/10.1089/adt.2016.701
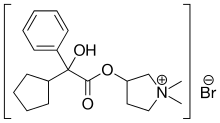
 Vernakalant
Vernakalant  Ibutilide
Ibutilide