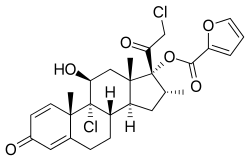
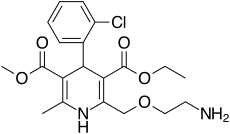
Takeda Pharmaceutical Company Limited (“Takeda”) announced the U.S. Food and Drug Administration (FDA) approval of Livtencity™ (maribavir) for the treatment of adults and pediatric patients (12 years of age or older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir, or foscarnet. Overall, more than twice the proportion of adult transplant patients with refractory or resistant (R/R) CMV infection/disease achieved confirmed CMV DNA level <LLOQ* (lower limit of quantification, i.e. <137 IU/mL) at Week 8 (end of treatment phase), the study’s primary endpoint, with Livtencity (56%; n=131/235), compared to those treated with conventional antiviral therapies (24%; n=28/117) (adjusted difference: 33%, 95% CI: 23–43; p<0.001). Livtencity is Takeda’s second new molecular entity to receive FDA approval in FY2021.

Livtencity is a new molecular entity which targets CMV at pUL97, resulting in inhibition of viral DNA replication, encapsidation and nuclear egress. Though a rare disease overall, CMV is one of the most common infections experienced by transplant recipients, with an estimated incidence rate of around 16%–56% in solid organ transplant (SOT) recipients3 and 30%–70% in hematopoietic stem cell (HSCT) transplant patients.2 CMV can be acquired or reactivated following transplant leading to serious consequences—including loss of the transplanted organ and failure of the graft—or loss of life. In patients with compromised immunity, CMV causes clinically challenging complications that can be fatal.Livtencity will be available in the coming days. For appropriate patients, physicians can submit a prescription to initiate access to treatment by contacting Takeda Patient Support at 1-855-268-1825.
“The FDA approval of Livtencity marks a major step forward in the treatment of post-transplant CMV, bringing a new therapeutic option to those living with this potential life-threatening opportunistic infection,” said Roy F. Chemaly, M.D., M.P.H., FACP, FIDSA, Department of Infectious Diseases, Infection Control & Employee Health at The University of Texas MD Anderson Cancer Center in Houston, TX. “In clinical studies, we observed Livtencity was statistically superior to conventional antiviral therapies in achieving the primary endpoint at Week 8.”
Prior to FDA approval, Livtencity (maribavir) was granted Orphan Drug Designation by the FDA for treatment of clinically significant CMV viremia and disease in at-risk patients, as well as Breakthrough Therapy Designation as a treatment for CMV infection and disease in transplant patients resistant or refractory to prior therapy. Takeda is looking forward to continuing our discussions with regulatory agencies across the globe to potentially bring maribavir to patients worldwide. The company is also investigating maribavir as a first-line treatment of CMV in hematopoietic stem cell transplant recipients in an ongoing Phase 3 clinical trial.
Livtencity was evaluated in the TAK-620-303 (SOLSTICE) trial, a global, multicenter, randomized, open-label, active-controlled superiority trial assessing the efficacy and safety of treatment with either maribavir or investigator-assigned treatment (IAT, conventional antiviral therapy) in 352 HSCT and SOT adult recipients with CMV infection refractory, with or without or resistance, to one or a combination of conventional antiviral therapies: ganciclovir, valganciclovir, foscarnet, or cidofovir. Participants were randomized 2:1 to receive maribavir (N=235) (400 mg, twice daily) or IAT (N=117) (as dosed by the investigator) for up to 8-weeks. After completion of the treatment period, subjects entered a 12-week follow-up phase. The primary efficacy endpoint was confirmed CMV DNA level <LLOQ* (lower limit of quantification, [i.e. <137 IU/mL] as assessed by COBAS® AmpliPrep/COBAS® TaqMan® CMV test at the end of Week 8).
The most common adverse events occurring in all grades, >10% of patients receiving maribavir were taste disturbance, nausea, diarrhea, vomiting, and fatigue. A higher proportion of subjects in the IAT group discontinued study medication due to an adverse event compared to the Livtencity group (32%, n=37/116 versus 13%, n=31/234, respectively). Taste disturbance events (46%, n=108/234) were generally mild, and rarely led to discontinuation of maribavir (1%). In 37% of patients, these events resolved while patients remained on therapy (median duration 43 days; range 7 to 59 days). For the patients with ongoing taste disturbance after drug discontinuation, resolution occurred in 89%.1 In patients with resolution of symptoms after drug discontinuation, the median duration of symptoms off treatment was 6 days (range 2 to 85 days). All-cause mortality was similar in each treatment group (Livtencity 11%, n=27/235; IAT 11%, n=13/117).
More..
https://en.wikipedia.org/wiki/Maribavir
https://go.drugbank.com/drugs/DB06234