
New treatment option combines proven efficacy and established safety profile of buprenorphine with a novel delivery system that adds convenience and flexibility.

Endo Pharmaceuticals Inc., a subsidiary of Endo International plc (NASDAQ: ENDP) (TSX: ENL), and BioDelivery Sciences International, Inc. (NASDAQ: BDSI), announced today that the U.S. Food and Drug Administration (FDA) has approved BELBUCA™ (buprenorphine) buccal film for use in patients with chronic pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate. BELBUCA™, which is the first and only buprenorphine developed with a dissolving film that is absorbed through the inner lining of the cheek for chronic pain management, is expected to be commercially available in the U.S. during the first quarter of 2016 in seven dosage strengths, allowing for flexible dosing ranging from 75 μg to 900 μg every 12 hours. This enables physicians to individualize titration and treatment based on the optimally effective and tolerable dose for each patient.
“The availability of new, convenient and flexible treatment options is important for patients whose lives are burdened by chronic pain, a debilitating condition that affects more Americans than diabetes, heart disease and cancer combined,” said Richard L. Rauck, M.D., Director of Carolinas Pain Institute, Winston Salem, NC. “BELBUCA™ provides a unique approach for chronic pain management, combining the proven efficacy and established safety of buprenorphine with a novel buccal film delivery system that adds convenience and flexibility. For both opioid-naïve and opioid-experienced patients who require around-the-clock treatment and for whom alternative treatment options are inadequate, BELBUCA™ offers appropriate, consistent pain relief and a low incidence of typical opioid-like side effects.”
BELBUCA™ is a mu-opioid receptor partial agonist and a potent analgesic with a long duration of action that utilizes BDSI’s patented BioErodible MucoAdhesive (BEMA®) drug delivery technology. Through this unique delivery system, buprenorphine is efficiently and conveniently delivered across the buccal mucosa (inside lining of the cheek). Buprenorphine is a Schedule III controlled substance, meaning that it has been defined as having lower abuse potential than Schedule II drugs, a category that includes most opioid analgesics. Among chronic pain patients taking opioids, the vast majority are on daily doses of 160 mg of oral morphine sulfate equivalent (MSE) or less. With seven dosage strengths up to 160 mg MSE, BELBUCA™ offers a treatment choice for a wide range of opioid needs in chronic pain sufferers.






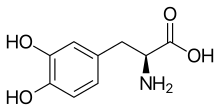



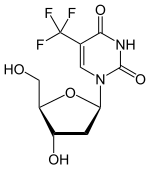 (Trifluridine)
(Trifluridine) 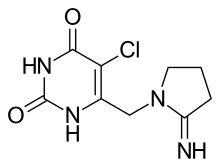 (Tipiracil)
(Tipiracil)









