Cancer drops sparse chemical hints of its presence early on, but unfortunately, many of them are in a class of biochemicals that could not be detected thoroughly, until now.
Researchers at the Georgia Institute of Technology have engineered a chemical trap that exhaustively catches what are called glycoproteins, including minuscule traces that have previously escaped detection.
Glycoproteins are protein molecules bonded with sugar molecules, and they're very common in all living things. Glycoproteins come in myriad varieties and sizes and make up important cell structures like cell receptors. They also wander around our bodies in secretions like mucus or hormones.
But some glycoproteins are very, very rare and can serve as an early signal, or biomarker, indicating there's something wrong in the body – like cancer. Existing methods to reel in glycoproteins for laboratory examination are relatively new and have had big holes in their nets, so many of these molecules, especially those very rare ones produced by cancer, have tended to slip by.
Cancerous traces
"These tiny traces are critically important for early disease detection," said principal investigator Ronghu Wu, a professor in Georgia Tech's School of Chemistry and Biochemistry. "When cancer is just getting started, aberrant glycoproteins are produced and secreted into body fluids such as blood and urine. Often their abundances are extremely low, but catching them is urgent."
This new chemical trap, which took Georgia Tech chemists several years to develop and is based on a boronic acid, has proven extremely effective in lab tests including on cultured human cells and mouse tissue samples.
"This method is very universal," said first author Haopeng Xiao, a graduate research assistant. "We get over 1,000 glycoproteins in a really small lab sample."
In comparison tests with existing methods, the chemical trap, a complex molecular construction reminiscent of an octopus, captured exponentially more glycoproteins, especially more of those trace glycoproteins.
Wu, Xiao and Weixuan Chen, a former Georgia Tech postdoctoral researcher, who was also first author of the study alongside Xiao, published their results in the journal Nature Communications. The research was funded by the National Science Foundation and the National Institutes of Health.
Boronic bungles
For chemistry whizzes, here's a short summary of how the researchers made the octopus. They took a good thing and doubled then tripled down on it.
Those who recall high school chemistry class may still know what boric acid is, as do people who use it to kill roaches. Its chemical structure is an atom of boron bonded with three hydroxyl groups (H3BO3).
Boronic acids are a family of organic compounds that build on boric acid. There are many members of the boronic acid family, and they tend to bond well with glycoproteins, but their bonds can be less reliable than needed.
"Most boronic acids let too many low-abundance glycoproteins get away," Wu said. "They can catch glycoproteins that are in high abundance but not those in low abundance, the ones that tell us more valuable things about cell development or about human disease."
Benzoboroxole octopus
But the Georgia Tech chemists were able to leverage the strengths of boronic acids to develop a glycoprotein capturing method that works exceptionally well.
First, they tested several boronic acid derivatives and found that one called benzoboroxole strongly bound with each sugar component on the glycopeptide. ("Peptide" refers to the basic chemical composition of a protein.)
Then they stitched many benzoboroxole molecules together with other components to form a "dendrimer," which refers to the resulting branch- or tentacle-like structure. The finished large molecule resembled an octopus ready to go after those sugar components.
In its middle, similarly positioned to an octopus's head, was a magnetic bead, which acted as a kind of handle. Once the dendrimer caught a glycoprotein, the researchers used a magnet to grab the bead and pull out their chemical octopus along with its ensnared glycopeptides (e.g. glycoproteins).
"Then we washed the dendrimer off with a low pH solution, and we had the glycoproteins analyzed with things like mass spectrometry," Wu said.
Cancer treatments?
The researchers have some ideas about how medical laboratory researchers could make practical use of the new Georgia Tech method to detect odd biomolecules emitted by cancer, such as antigens. For example, the chemical octopus could improve detection of prostate-specific antigens (PSA) in prostate cancer screenings.
"PSA is a glycoprotein. Right now, if the level is very high, we know that the patient may have cancer, and if it's very low, we know cancer is not likely," Wu said. "But there is a gray area in between, and this method could lead to much more detailed information in that gray area."
The researchers also believe that developers could leverage the chemical invention to produce targeted cancer treatments. Immune cells could be trained to recognize the aberrant glycoproteins, track down their source cancer cells in the body and kill them.
The research's potential for science goes far beyond its possible future medical applications.
The fields of genomics and proteomics have made great strides. Following in their footsteps, this new molecular trap could advance the study of the rising field of glycoscience.
Source:http://www.rh.gatech.edu/news/605861/chemical-octopus-catches-sneaky-cancer-clues-trace-glycoproteins



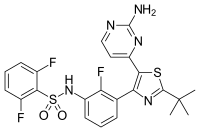



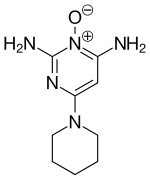 Finasterite
Finasterite 
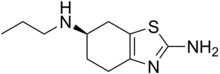
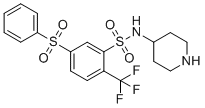 WAY-316606
WAY-316606