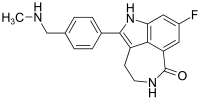
The metabolite of a drug that is helping patients battle multiple sclerosis appears to significantly slow the onset of Parkinson's disease, researchers say.
The oral drug, dimethylfumarate, or DMF, and its metabolite, monomethylfumarate, or MMF, both increase activity of Nrf2, a protein that helps protect the body from oxidative stress and inflammation, hallmarks of both diseases, said Dr. Bobby Thomas, neuroscientist in the Department of Pharmacology and Toxicology at the Medical College of Georgia at Augusta University.
 dimethylfumarate
dimethylfumarate  monomethylfumarate,
monomethylfumarate,
But the new study provides the first evidence that the metabolite, which is essentially the active portion of the parent drug, more directly targets Nrf2, potentially reducing known side effects of the parent drug that include flushing, diarrhea, nausea, vomiting, abdominal pain and the brain infection encephalopathy, said Thomas, corresponding author of the study in The Journal of Neuroscience.
Particularly, the gastrointestinal side effects can exacerbate some problems patients with Parkinson's already experience, said Dr. John Morgan, neurologist, neuroscientist and Parkinson's disease specialist in the MCG Department of Neurology. In addition to destroying neurons in the brain that produce dopamine, a neurotransmitter that enables movement and learning, Parkinson's causes nerve cell death in the gastrointestinal tract and related problems such as severe constipation.
"Nrf2 is a natural protective mechanism we have for oxidative stress," Thomas said. The fact that multiple sclerosis and Parkinson's have in common evidence of declining activity of the Nrf2 pathway has generated interest in the drug for Parkinson's and other neurodegenerative diseases.
DMF was approved for multiple sclerosis three years ago by the Food and Drug Administration. While its metabolite MMF is not quite as potent as the parent drug in increasing Nrf2 activity, the new study indicates that its action is sufficient to dramatically slow the loss of dopamine-producing neurons as well as the parent drug, in an animal model of Parkinson's.
In their model, mice given the neurotoxin MPTP experience a dramatic loss of dopamine-producing neurons, losing about half within a handful of days, and rapidly develop Parkinson's-like symptoms. Patients, on the other hand, slowly develop symptoms over many years. By the time they seek medical care, patients may have lost 30-50 percent of their dopaminergic neurons, said Morgan, a study coauthor. "Presentation is after the disease is kind of out of the gate."
To accommodate the very compressed timeline in their model and the fact that several daily doses are needed before the drug starts to work, the researchers first gave the mice either the drug or metabolite the day before they started the toxin.
Dopamine-producing neurons are located in a darker-pigmented central portion of the brain called the substantia nigra. Even in the absence of disease, making dopamine is a stressful job for these neurons that makes them generally more fragile and actually results in oxidative stress even in a healthy scenario, Morgan said. To make a difficult situation worse, increased oxidative stress can make dopamine toxic to neurons, he said.
To increase Nrf2 activity, the parent drug DMF also appears to first make bad matters worse. DMF increases oxidative stress by depleting the natural antioxidant, glutathione, and reduces the power of cell powerhouses, called mitochondria, by limiting their ability to use oxygen and glucose to make energy leading to reduced viability of dopamine-producing cells, Thomas said.
The metabolite MMF appears to more directly activate Nrf2, and actually increases glutathione and improves mitochondrial function, brain cell studies showed. While the parent drug ultimately produces a higher Nrf2 activation, the researchers found the MMF effect was sufficient to stop the dramatic neuron loss in the animal model.
Both DMF and MMF slowed neuron loss to a more normal level, and the neurons that survived continued to make dopamine. Inflammation and oxidative stress levels also were significantly reduced, the researchers said.
As a next step, they are working toward a clinical trial of MMF in patients with early Parkinson's disease. Although the metabolite could be easily formulated for humans, it has not yet been done, Thomas notes.
"If we can catch them early enough, maybe we can slow the disease," Morgan said. "If it can help give five to eight more years of improved quality of life that would be great for our patients."
Clinical studies of the drug in Parkinson's are being planned in the United Kingdom and additional analogues of its metabolite, which could be used clinically and which the researchers think ultimately will be the best option for patients, are under development.
Oxidative stress is a byproduct of the body's use of oxygen. Free radicals, generated by oxygen use, are unstable molecules that can interfere with usual cell function and are believed to contribute to a wide range of conditions from normal aging to Alzheimer's disease. Simply giving antioxidants, such as vitamin E, which work more like scavengers to scarf up free radicals, has not worked in combating neurodegenerative disease, Thomas said. He's optimistic that directly targeting Nrf2 will be effective in at least slowing the disease, but there remains a need for clinically safe Nrf2 activators.
Activity of the Nrf2 pathway tends to slowly decline with age. Exercise upregulates Nrf2, and Morgan regularly encourages his patients to be as active as possible. A small group of patients with Parkinson's in Europe has a concentrated activation of Nrf2 that at least delays their disease onset. Parkinson's tends to be diagnosed in the mid-to-late 50s and early 60s and is more common in men.
One concern with chronically elevating anti-oxidant and anti-inflammatory molecules with drugs like DMF and MMF is creating some of the same problems that immunosuppressive drugs given to organ transplant patients create. Chronic suppression of the immune response makes patients more susceptible to invaders like cancers and infections.
Ref : http://www.jneurosci.org/content/36/23/6332.short?sid=e7a934c5-6996-4fff-bfa9-f2c600507e1f






 Valsartan
Valsartan