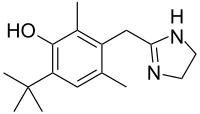
In continuation of my update on Everolimus
Novartis today announced that the United States Food and Drug Administration (FDA) approved Afinitor® (everolimus) tablets for the treatment of adult patients with progressive, well-differentiated, nonfunctional neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin that are unresectable, locally advanced or metastatic. Afinitor received a priority review designation providing a shortened review period for drugs that treat serious conditions and offer a significant improvement in safety or effectiveness.
"Afinitor is the first treatment approved for progressive, nonfunctional NET of lung origin, and one of very few options available for progressive, nonfunctional GI NET, representing a shift in the treatment paradigm for these cancers," said Bruno Strigini, President, Novartis Oncology. "We are proud of our Afinitor development program, which has translated to meaningful benefits for patients with several different cancers and rare diseases."
Neuroendocrine tumors are a rare type of cancer that originate in neuroendocrine cells throughout the body, and are most often found in the GI tract, lungs or pancreas. NET can be defined as functional or nonfunctional. Functional NET are characterized by symptoms caused by the oversecretion of hormones and other substances. Nonfunctional NET may be characterized by symptoms caused by tumor growth, such as intestinal obstruction, pain and bleeding for GI NET, and asthma, chronic obstructive pulmonary disease and pneumonia for lung NET. More than 70% of patients with NET have nonfunctional tumors. At the time of diagnosis, 5%-44% (depending on site of tumor origin) of patients with NET in the GI tract and 28% of patients with lung NET have advanced disease, meaning the cancer has spread to other areas of the body, making it difficult to treat. Progression, or the continued growth or spread of the tumor, is typically associated with poor outcomes.
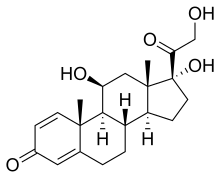 (Prednisole)
(Prednisole) (Indomethacin)
(Indomethacin)
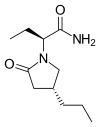
 (
( (Donepezil)
(Donepezil) 

 (Elbasvir)
(Elbasvir)  (Grazoprevir)
(Grazoprevir)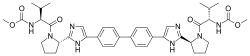
 Tetracaine
Tetracaine