
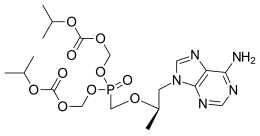
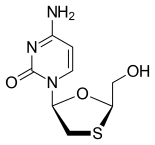
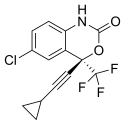
Tenofovir disoproxil fumarate Lamivudine Efavirenz
In continuation of my update, on Tenofovir, Lamivudine and Efavirenz
Global pharmaceutical company Mylan N.V. announced that it will introduce in the U.S. a third cost-saving HIV combination. The U.S. Food and Drug Administration (FDA) approved Symfi (efavirenz, lamivudine and tenofovir disoproxil fumarate) 600 mg/300 mg/300 mg tablets, a once-daily, single-tablet regimen (STR), indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adult and pediatric patients weighing at least 40 kg.
"As the largest supplier of antiretrovirals by volume in the world, Mylan has a longstanding commitment to expanding affordable access to treatments for people living with HIV," said Mylan CEO Heather Bresch. "As we continue to grow our U.S. portfolio of ARV products, now including Symfi Lo™, Symfi™, and Cimduo™, we are providing access to patients and empowering them to choose the lower-cost ARV treatment option that is right for them."
The introduction of Symfi™ comes after the FDA's recent approval of two Mylan ARVs: Cimduo™ (lamivudine and tenofovir disoproxil fumarate) 300 mg/300 mg tablets, a once-daily combination of two nucleo(t)side reverse transcriptase inhibitors, which is indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 35 kg.; and Symfi Lo™ (efavirenz, lamivudine and tenofovir disoproxil fumarate) 400 mg/300 mg/300 mg tablets, also approved for patients with HIV-1 in adults and pediatric patients weighing at least 35 kg.
Following FDA approval, Mylan launched Symfi Lo™ earlier in March. It expects Cimduo™ and Symfi™ to launch in the second quarter of 2018.
Symfi™ and Symfi Lo™ feature the same triple combination of molecules; however, Symfi Lo™ features a reduced dose of efavirenz while Symfi™ uses a dosing similar to other efavirenz products already on the market. The combination represented by Symfi™ (efavirenz, lamivudine and tenofovir disoproxil fumarate) 600 mg/300 mg/300 mg tablets is the most widely-taken ARV regimen outside of the U.S., with more than 7 million users worldwide in 20161.
In 2017, HIV was the category with highest pharmacy spend for Medicaid, the third highest for health exchange plans and the fifth highest for commercial plans.2 According to IQVIA, total spending on HIV drugs has more than tripled since 2007, outpacing the approximate 60% growth in overall drug spending.
To help reduce the high cost of HIV treatment in the U.S, the list price of these Mylan ARVs will be discounted significantly from the wholesale acquisition cost of similar medicines on the market.
"Mylan has been on the forefront of bringing innovative delivery and dosage forms of ARVs to millions of patients in the developing world," said Mylan President Rajiv Malik. "We've already extended our reach to people in the U.S. living with HIV with the introduction of Symfi Lo™ and Cimduo™. Adding Symfi™ to our portfolio further strengthens our commitment to investing in developing and manufacturing these important products."
Lactic acidosis and hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues including lamivudine and tenofovir disoproxil fumarate. Severe acute exacerbations of hepatitis B have been reported in patients who are co-infected with hepatitis B virus and HIV who have discontinued lamivudine and tenofovir disoproxil fumarate.
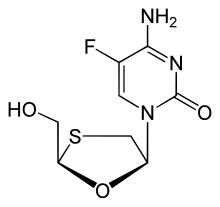 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide










{kind=link}