In continuation of my update on meloxicam,
Axsome Therapeutics, Inc. that the U.S. Food and Drug Administration (FDA) has accepted for filing the Company’s New Drug Application (NDA) for AXS-07 for the acute treatment of migraine, and has set a Prescription Drug User Fee Act (PDUFA) target action date of April 30, 2022 for the NDA. AXS-07 (MoSEIC™ meloxicam-rizatriptan) is a novel, oral, rapidly absorbed, multi-mechanistic, investigational medicine for migraine.
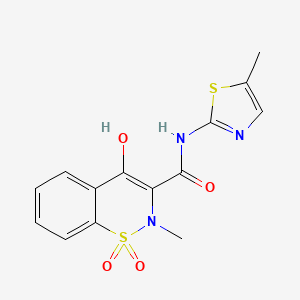
meloxicam
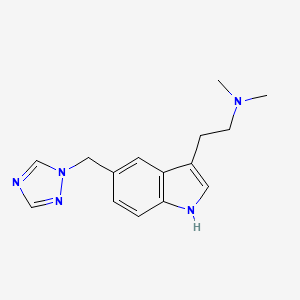
Rizatriptan
“The FDA’s acceptance of the NDA for AXS-07 is an important milestone for Axsome as it brings us closer to potentially making this multi-mechanistic treatment available to migraine patients in need,” said Herriot Tabuteau, MD, Chief Executive Officer of Axsome. “We look forward to continued interactions with the FDA during the review process.”
The NDA is supported by results from two Phase 3 randomized, double-blind, controlled trials of AXS-07 in the acute treatment of migraine, the MOMENTUM and INTERCEPT trials, which demonstrated statistically significant elimination of migraine pain with AXS-07 compared to placebo and active controls.
AXS-07 is a novel, oral, rapidly absorbed, multi-mechanistic investigational medicine for the acute treatment of migraine, consisting of MoSEIC™ meloxicam and rizatriptan. Meloxicam is a new molecular entity for migraine enabled by Axsome’s MoSEIC (Molecular Solubility Enhanced Inclusion Complex) technology, which results in rapid absorption of meloxicam while maintaining a long plasma half-life. Meloxicam is a COX-2 preferential non-steroidal anti-inflammatory drug and rizatriptan is a 5-HT1B/1D agonist. AXS-07 is designed to provide rapid, enhanced and consistent relief of migraine, with reduced symptom recurrence. AXS-07 is covered by more than 80 issued U.S. and international patents which provide protection out to 2036. AXS-07 is not approved by the FDA.
https://pubchem.ncbi.nlm.nih.gov/compound/Rizatriptan
https://pubchem.ncbi.nlm.nih.gov/compound/Meloxicam#section=2D-Structure