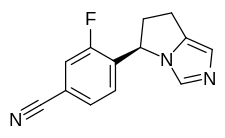
Myers Squibb Company (NYSE: BMY) announced that the U.S. Food and Drug Administration (FDA) approved Zeposia (ozanimod) 0.92 mg for the treatment of adults with relapsing forms of multiple sclerosis (RMS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease.1 Zeposia, an oral medication taken once daily, is the only approved sphingosine-1-phosphate (S1P) receptor modulator that offers RMS patients an initiation with no genetic test and no label-based first-dose observation required for patients.1,4,5 An up-titration scheme should be used to reach the maintenance dosage of Zeposia, as a transient decrease in heart rate and atrioventricular conduction delays may occur.

Multiple sclerosis (MS) is a disease in which the immune system attacks the protective myelin sheath that covers the nerves, creating damaging lesions that make it harder for signals to travel between each nerve cell.6,7 This “signal breakdown” can lead to symptoms and relapses.6,8
“With the FDA approval of Zeposia, appropriate patients with relapsing forms of multiple sclerosis will have another oral treatment option with meaningful efficacy to help address the disease’s hallmark relapses and brain lesions,”9 said Samit Hirawat, M.D., chief medical officer, Bristol Myers Squibb. “Zeposia has substantial clinical potential, and we are well positioned with our heritage in transformational science to ensure this innovative compound ultimately benefits as many patients as possible.”
The approval is based on data from the largest pivotal, head-to-head RMS studies with an active comparator to date: the randomized, active-controlled Phase 3 SUNBEAM™ (safety and efficacy of Zeposia versus interferon beta-1a in relapsing multiple sclerosis) and RADIANCE™ (safety and efficacy of the selective sphingosine 1-phosphate receptor modulator Zeposia in relapsing multiple sclerosis) Part B clinical trials of more than 2,600 adults.1,2,3,10 In both trials – as compared to AVONEX® (interferon beta-1a), Zeposia delivered powerful efficacy as measured by annualized relapse rate (ARR), as well as on the number and size of brain lesions.1,2,3
- Zeposia demonstrated a relative reduction in ARR versus AVONEX of 48% through one year and 38% at two years (absolute ARR of 0.18 versus 0.35 and 0.17 versus 0.28, respectively).1,2,3
- At one year, treatment with Zeposia reduced the number of T1-weighted gadolinium-enhanced (GdE) brain lesions more than AVONEX (0.16 vs 0.43), a relative reduction of 63%, and reduced the number of new or enlarging T2 lesions (1.47 vs. 2.84), a relative reduction of 48%.1,3
- At two years, treatment with Zeposia reduced the number of T1-weighted gadolinium-enhanced (GdE) brain lesions more than AVONEX (0.18 vs 0.37), a relative reduction of 53%.1,2 Zeposia also reduced the number of new or enlarging T2 lesions vs AVONEX (1.84 vs 3.18), a relative reduction of 42%.1,2
There was no statistically significant difference in the three-month and six-month confirmed disability progression between Zeposia- and AVONEX- treated patients over two years.1
Zeposia demonstrated acceptable safety and tolerability in the Phase 3 SUNBEAM and RADIANCE Part B trials.1,2,3 Zeposia is contraindicated in patients who in the last six months experienced myocardial infarction, unstable angina, stroke, transient ischemic attack (TIA), decompensated heart failure requiring hospitalization, or Class III/IV heart failure; patients who have a presence of Mobitz type II second or third-degree atrioventricular (AV) block, sick sinus syndrome, or sino-atrial, unless the patient has a functioning pacemaker; patients with severe untreated sleep apnea; and patients taking a monoamine oxidase inhibitor.1 Zeposia is associated with the following Warnings and Precautions: increased risk of infections, bradyarrhythmia and atrioventricular conduction delays, liver injury, fetal risk, increased blood pressure, respiratory effects, macular edema, posterior reversible encephalopathy syndrome, additive immunosuppressive effects from prior immune-modulating treatments, severe increase in disability after stopping Zeposia, and immune system effects after stopping Zeposia.1 Please see Important Safety Information for additional details. The most common adverse reactions (incidence ≥4%) were upper respiratory infection, hepatic transaminase elevation, orthostatic hypotension, urinary tract infection, back pain, and hypertension.1
Before initiation of treatment with Zeposia, all patients require assessments including a recent complete blood count including lymphocyte count (within six months or after discontinuation of prior MS therapy), an ECG to determine whether preexisting conduction abnormalities are present, a recent liver function test (within six months), and consideration of current and prior medications, including vaccinations.1 For patients with a history of uveitis or macular edema, an ophthalmic assessment is required.
Treatment for relapsing forms of multiple sclerosis is critical to address this devastating neurological disease.11 I’m excited, with the introduction of Zeposia, I will have a new oral option to offer my RMS patients that has demonstrated efficacy and safety,”1 said Bruce Cree, M.D., Ph.D., M.A.S., professor of clinical neurology, University of California San Francisco (UCSF) Weill Institute for Neurosciences and clinical research director, UCSF MS Center.
“Multiple sclerosis is an unpredictable and often disabling disease that affects nearly one million people in the United States.9,12 Ongoing treatment with disease-modifying therapy can reduce the number of disease attacks,”11 said Bruce Bebo, executive vice president of research, National Multiple Sclerosis Society. “Each person can respond differently to these medications, which is why having treatment options is so important. We are pleased that there will now be another effective treatment option for people with MS.”
As the country’s healthcare system is dealing with the unprecedented COVID-19 pandemic, Bristol Myers Squibb has made the decision to delay commercialization of Zeposia. The Company made the decision based on what’s in the best health interest of our patients, customers and employees. Bristol Myers Squibb will continue to monitor the environment and will partner with the neurology community to inform launch timing.
A Marketing Authorization Application for Zeposia for the treatment of adults with relapsing-remitting multiple sclerosis in the European Union is currently under review with the European Medicines Agency (EMA). A regulatory decision from the EMA is expected in the first half of 2020.
https://en.wikipedia.org/wiki/Ozanimod
FDA Approves Zeposia (ozanimod) for Relapsing Forms of Multiple Sclerosis