
In continuation of my update on mifepristone
A combination of the drugs mifepristone and misoprostol can help bring closure to some women and their families suffering from miscarriage, and reduces the need for surgical intervention to complete the painful miscarriage process. Results of a new clinical trial led by researchers at the Perelman School of Medicine at the University of Pennsylvania, show that while the standard drug regimen using misoprostol on its own frequently fails to complete the miscarriage, a combination of misoprostol and the drug mifepristone works much more reliably. The report is published today in the New England Journal of Medicine.
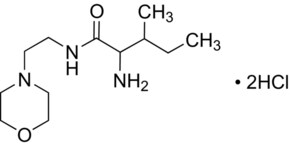
 mifepristone
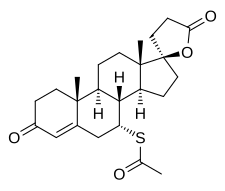
mifepristone 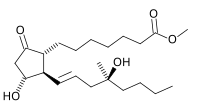 misoprostol
misoprostol
Each year in the United States alone approximately 1 million women have miscarriages. When the body does not expel the pregnancy tissue on its own – the final part of a miscarriage – women need to undergo a surgical procedure or take the drug misoprostol. Though often preferable for its convenience and privacy – patients can take it in the comfort of their own homes – misoprostol does not always work, and many women who use misoprostol are still left with no option but to undergo an invasive procedure they wished to avoid, prolonging an already physically and emotionally difficult situation.
"Though rarely discussed openly, miscarriage is the most common complication of pregnancy, and the public health burden is both physical and psychological. For too many women, misoprostol alone just leads to frustration. I have seen my patients suffer from the insult of the treatment failure added to the injury of the initial loss," said study lead author Courtney A. Schreiber, MD, MPH, chief of the division of Family Planning and an associate professor of Obstetrics and Gynecology at the Perelman School of Medicine at the University of Pennsylvania. "As physicians, we have to do better for these patients, and our new study shows that by combining mifepristone with misoprostol, we can."
Mifepristone is used along with misoprostol to induce abortion in early pregnancy. But the effectiveness of mifepristone-misoprostol for miscarriage patients, in comparison to the commonly used misoprostol alone, has been unclear.
In the new study, 300 women who had been diagnosed with early pregnancy loss – described as a miscarriage in the first trimester – were assigned to receive the standard 800 micrograms of misoprostol placed vaginally. Half were also randomly assigned to receive pretreatment with a 200 mg pill of mifepristone, which primes the uterus to respond to misoprostol's contraction-inducing effect.
The researchers found that overall, 91.2 percent of women receiving the mifepristone pretreatment plus misoprostol experienced gestational sac expulsion -; the definition of a completed miscarriage -; 83.8 percent by their first follow-up visit, which occurred two days after the treatment on average. Misoprostol alone was only effective 75.8 percent of the time, with 67.1 percent completing by their first follow up visit.
The researchers looked at a variety of outcomes in the study, and essentially all were better for the women taking mifepristone plus misoprostol. Patients assigned to this group, for example, had a much lower chance (8.8 percent) of needing a surgical intervention by day 30, compared to 23.5 percent for the misoprostol-alone group. There were no significant differences between the groups in terms of pain, bleeding, or other side-effects. Notably, serious side effects were rare in both groups.
Mifepristone is a highly regulated medication. At present, the U.S. Food and Drug Administration requires that the drug be dispensed only in registered hospitals, clinics and doctor's offices, but not in retail pharmacies. Schreiber says that physicians who wish to treat women with miscarriages – including but not limited to physicians in obstetrics, internal medicine, emergency medicine, and family medicine – should consider registration.
"High-quality care for women who suffer miscarriage not only improves physical outcomes, but helps alleviate the psychosocial stress that can accompany the loss of a pregnancy," she says. "Given how common miscarriage is and the effectiveness of the drug combination as shown in this new study, any doctor who cares for women who become pregnant, and therefore could have a miscarriage, should be registered to prescribe and dispense mifepristone."
Ref : https://www.pennmedicine.org/news/news-releases/2018/june/drug-combination-offers-more-effective-care-for-patients-suffering-miscarriage-penn-study-shows




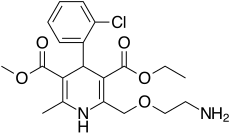
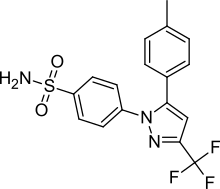




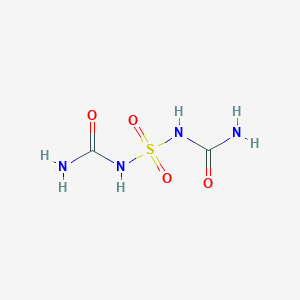
 S
S