We know that, Buprenorphine/samidorphan (developmental code name ALKS-5461) is a combination drug formulation of buprenorphine and samidorphan acting as a μ-opioid receptor antagonist which is under development by Alkermes as an adjunct to antidepressant therapy in treatment-resistant depression (TRD).
Buprenorphine
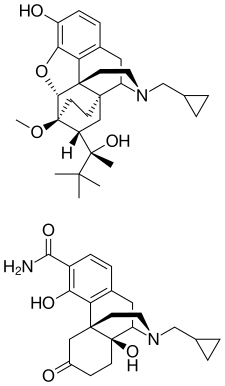 Samidorphan
Samidorphan
Alkermes plc (Nasdaq: ALKS)announced that the U.S. Food and Drug Administration (FDA) has accepted for review the New Drug Application (NDA) for ALKS 5461, a novel, once-daily, oral investigational medicine for the adjunctive treatment of major depressive disorder (MDD) in patients with an inadequate response to standard antidepressant therapies. FDA's target action date for the ALKS 5461 NDA is Jan. 31, 2019.
FDA's acceptance of the ALKS 5461 NDA and rescission of the Refusal to File letter issued March 30, 2018 follows productive interactions with the Agency in which Alkermes clarified certain aspects of the NDA submission. No additional data or analyses were submitted by Alkermes to FDA.
"FDA's filing of the ALKS 5461 application is a positive step forward for patients suffering from major depressive disorder, a serious disease where inadequate response to existing antidepressants remains a well-known and significant treatment limitation, and where there have been no new pharmacological treatment approaches in 30 years," stated Craig Hopkinson, M.D., Chief Medical Officer and Senior Vice President of Medicines Development and Medical Affairs at Alkermes. "We will continue to engage with the FDA throughout the review process, as we work to bring this important medicine to patients."
The NDA filing for ALKS 5461 is based on results from a clinical efficacy and safety package with data from more than 30 clinical trials and more than 1,500 patients with MDD. Throughout the clinical development program, ALKS 5461 demonstrated a consistent profile of antidepressant activity, safety and tolerability in the adjunctive treatment of MDD.