
Cempra, Inc. , a clinical-stage pharmaceutical company focused on developing antibiotics to meet critical medical needs in the treatment of bacterial infectious diseases, announced the completion of its rolling submission of the New Drug Applications (NDA) for solithromycin to the U.S. Food and Drug Administration (FDA) for the treatment of community-acquired bacterial pneumonia (CABP). Based on the Qualified Infectious Disease Product (QIDP) designation by the FDA of solithromycin, Cempra has Priority Review and has been granted Fast Track for both the oral capsule and intravenous formulations for the treatment of CABP, which could result in an FDA decision on solithromycin's NDA within eight months, or by the end of 2016, based on the Prescription Drug User Fee Act (PDUFA) performance goals.
 Solithromycin
Solithromycin
"Completion of the rolling submission of our first NDAs during Cempra's ten year anniversary year represents a major milestone for the company and a significant step toward our goal of developing antibiotics to meet the critical medical needs of patients in the treatment of bacterial infectious diseases," stated Prabhavathi Fernandes, Ph.D., president and chief executive officer of Cempra. "We believe the intravenous and capsule formulations will provide dosing flexibility that could lead to fewer hospital admissions, earlier discharge if admitted, and increased treatment of CABP on an outpatient basis. We are confident we have a strong data package for solithromycin."
"The management of CABP remains a challenge to healthcare professionals and I firmly believe that solithromycin has the potential to be a significant part of the treatment of this life threatening illness, given its published clinical efficacy and potential for multiple formulations," stated Thomas M. File, M.D., principal investigator for solithromycin clinical trials, Northeast Ohio Medical University. "Solithromycin's potency, spectrum of activity and tolerability could help to offset the rising problem of bacterial resistance, and it is gratifying to note that patients could be closer to benefiting from this potential new treatment."
The FDA has a 60-day filing review period to determine whether the NDAs are complete and acceptable for filing, and to confirm that Priority Review has been granted. Cempra expects to communicate the agency's decision regarding acceptance of the NDAs and its PDUFA date when it is known. Cempra's submissions in the EU remain on track for completion by the end of June 2016.
About Solithromycin
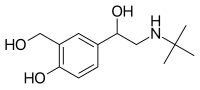
Solithromycin is a highly potent next-generation macrolide, the first fluoroketolide, which has potent activity against most macrolide-resistant strains. In vitro and in vivo studies have shown potent activity against S. pneumoniae as well as an extended spectrum of activity against CA-MRSA, streptococci, Haemophilus, enterococci, Mycobacterium avium and in animal models of malaria. It is also active against atypical bacteria, such as legionella, chlamydia, mycoplasma and ureaplasma, and against gonococci and other organisms that cause genitourinary tract infections. It is 8-16 times more potent than azithromycin against many bacteria and is active against azithromycin-resistant strains. Solithromycin's activity against resistant strains is driven by its ability to interact with three sites on the bacterial ribosome, compared to one for current macrolides. The binding to bacterial ribosomes and interaction with three ribosomal sites is expected to limit the development of bacterial resistance to solithromycin.

 promethazine
promethazine






 Valsartan
Valsartan

