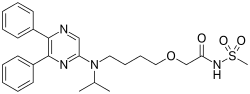
Living with pulmonary arterial hypertension is challenging, but the chore of treating the rare heart disease may change following promising clinical trial data to be published in the Dec. 24 issue of the New England Journal of Medicine.
Data from the largest study ever of pulmonary hypertension shows the oral medication Selexipag led to a 40 percent reduction in hospitalizations and worsening symptoms among patients with pulmonary hypertension.
Selexipag targets a well-known disease pathway that opens blood vessels to the lungs and improves heart function and is easier to use than PH treatments delivered with infusions or injections.
"For more than two decades we've targeted the prostacyclin pathway to induce vasodilation in these blood vessels in the lung," says senior study author Vallerie McLaughlin, M.D., director of the University of Michigan Pulmonary Hypertension Program.
"But because of the cumbersome nature of treatment, patients would often wait until late stages to begin therapy. Having an oral medication to attack the disease pathway will be a major advancement because less ill patients will be willing to begin this therapy," McLaughlin says.
Selexipag demonstrated effectiveness while its most frequent adverse events were headache, diarrhea, nausea, muscle pain and joint pain. The side effects were considered consistent with prostacyclin therapy.
The medication is not commercially available. Following the phase 3 clinical trial, the U.S. Food and Drug Administration is evaluating the drug.
Pulmonary hypertension, which is high blood pressure in the loop of blood vessels connecting the heart and lungs, can make everyday activities exhausting. It often leads to life-threatening heart failure as the heart works harder to pump blood to the lungs.
The greatest number of cases is reported in women between ages 21 and 40 who most often experience fatigue and shortness of breath.
While a rare disease, research of causes and treatment of pulmonary hypertension is growing. The GRIPHON study is a standout because of its size and scope, enrolling 1,156 patients with pulmonary hypertension from 181 centers from 39 countries in North and South America, Europe, Asia-Pacific and Africa.