
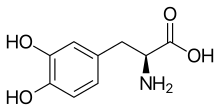
In continuation of my update on L-DOPA
A drug already used safely to treat Parkinson's disease, restless leg syndrome and other movement disorders also could delay or prevent the most common cause of blindness affecting more than 9 million older Americans - age-related macular degeneration (AMD).
Researchers have discovered that patients who take the drug L-DOPA are significantly less likely to develop AMD, and if they do get AMD it's at a significantly older age, according to the study published online Nov. 4 in the American Journal of Medicine. The retrospective study was led by researchers at Marshfield Clinic Research Foundation, University of Arizona, Medical College of Wisconsin, University of Miami, Essentia Health, Stanford University and University of Southern California.
"Research points to this as a pathway to regulate and prevent this most common cause of blindness in adults," said Murray Brilliant, Ph.D., director, Marshfield Clinic Research Foundation Center for Human Genetics, Marshfield, Wisconsin. "Imagine telling patients we potentially have medication that will allow them to see and continue enjoying life, their family and perform every day activities as they age. That is very powerful."
AMD, the No. 1 cause of legal blindness in adults over 60, is a progressive eye condition affecting as many as one in three adults. The disease attacks the macula of the eye, where the sharpest central vision occurs, causing central blindness. This vision is used to drive, read, recognize faces and perform daily tasks. AMD spares the peripheral vision, leaving dim images or black holes at the center of vision.
L-DOPA is a natural by-product of pigmentation and is made in a layer of cells in the back of the eye that functions to promote health and survival of retinal tissues. Researchers asked the question if people taking L-DOPA as a medicine are protected from AMD.
"The obvious question was if the L-DOPA no longer produced was supplemented via pill form, does it have the potential to serve as a preventive medicine against AMD," Brilliant said. "We need more research, but this first step is promising."



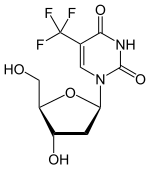 (Trifluridine)
(Trifluridine) 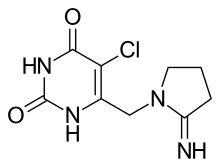 (Tipiracil)
(Tipiracil)




