In continuation of my update on tezacafto
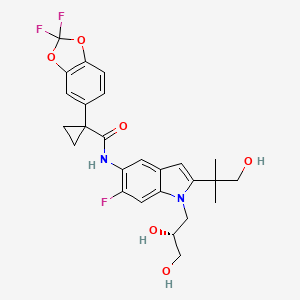
 Tezacaftor
Tezacaftor 
Lumacaftor/ivacaftor
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) announced the U.S. Food and Drug Administration (FDA) approved Symdeko (tezacaftor/ivacaftor and ivacaftor) for use in children with cystic fibrosis ages 6 through 11 years who have two copies of the F508del-CFTR mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that is responsive to Symdeko. It was previously approved by the FDA for use in patients with cystic fibrosis 12 years and older with two copies of the F508del mutation or one copy of a responsive mutation in the U.S. An additional dosage strength of Symdeko tablets is now available (tezacaftor 50 mg/ivacaftor 75 mg and ivacaftor 75 mg) in connection with this approval.
“Today’s expanded approval of Symdeko in children ages 6 through 11 is an important step in our efforts to continue to bring treatment options to the youngest patients possible and importantly brings us closer to our goal of developing medicines for all people living with CF,” said Reshma Kewalramani, M.D., Executive Vice President and Chief Medical Officer at Vertex.
Vertex completed a 24-week Phase 3 open-label, multicenter study to evaluate the pharmacokinetics, safety, and tolerability of tezacaftor/ivacaftor and ivacaftor in children ages 6 through 11 years in the U.S. and Canada. The regimen was generally well tolerated, and safety data were similar to what was observed in previous studies of patients aged 12 years and older. The full data from this study will be published later this year.
“We’ve seen the clinical impact of Symdeko in people with CF aged 12 years and above, and this approval marks a crucial milestone for patients ages 6 through 11 years who may benefit from CFTR modulation, enabling us to treat the basic defect in CF at an earlier stage of disease,” said Seth Walker, M.D., University Hospitals of Cleveland, Cleveland Medical Center, Rainbow Babies and Children’s Hospital. “Symdeko is an important treatment option for eligible people with CF who either never started or have discontinued another CFTR modulator.”
About Cystic Fibrosis
Cystic Fibrosis (CF) is a rare, life-shortening genetic disease affecting approximately 75,000 people in North America, Europe and Australia.
CF is caused by a defective or missing cystic fibrosis transmembrane conductance regulator (CFTR) protein resulting from mutations in the CFTR gene. Children must inherit two defective CFTR genes — one from each parent — to have CF. There are approximately 2,000 known mutations in the CFTR gene. Some of these mutations, which can be determined by a genetic test, or genotyping test, lead to CF by creating non-working or too few CFTR proteins at the cell surface. The defective function or absence of CFTR protein results in poor flow of salt and water into and out of the cell in a number of organs. In the lungs, this leads to the buildup of abnormally thick, sticky mucus that can cause chronic lung infections and progressive lung damage in many patients that eventually leads to death. The median age of death is in the mid-to-late 20s.
About Symdeko (tezacaftor/ivacaftor and ivacaftor)
Some mutations result in CFTR protein that is not processed or folded normally within the cell, and that generally does not reach the cell surface. Symdeko is a combination of tezacaftor and ivacaftor. Tezacaftor is designed to address the trafficking and processing defect of the CFTR protein to enable it to reach the cell surface where ivacaftor can increase the amount of time the protein stays open.
U.S. INDICATION FOR SYMDEKO® (tezacaftor/ivacaftor and ivacaftor) tablets
Symdeko is a prescription medicine used for the treatment of cystic fibrosis (CF) in patients aged 6 years and older who have two copies of the F508del mutation, or who have at least one mutation in the CF gene that is responsive to treatment with Symdeko. Patients should talk to their doctor to learn if they have an indicated CF gene mutation. It is not known if Symdeko is safe and effective in children under 6 years of age.
https://en.wikipedia.org/wiki/Tezacaftor
https://en.wikipedia.org/wiki/Lumacaftor/ivacaftor