
A compound discovered and developed by a team of Georgetown Lombardi Comprehensive Cancer Center researchers that halts cancer in animals with Ewing sarcoma and prostate cancer appears to work against some forms of leukemia, too. That finding and the team's latest work was published online Oct. 8 in Oncotarget.

The compound is YK-4-279, the first drug targeted at similar chromosomal translocations found in Ewing sarcoma, prostate cancer and in some forms of leukemia. Translocations occur when two normal genes break off from a chromosome and fuse together in a new location. This fusion produces new genes that manufacture proteins, which then push cancer cells to become more aggressive and spread. One of those proteins is EWS-FLI1. YK-4-279 appears effective in controlling the cancer promoting functions of EWS-FLI1.
"EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma, which occurs predominantly in children, teens and young adults," says Aykut Üren, MD, professor of molecular oncology at Georgetown Lombardi. "It also appears to drive cancer cell growth in some prostate cancers."
In this new study led by Üren, mice with EWS-FLI1-driven leukemia were given injections of YK-4-279 five days per week for two weeks and compared with untreated mice. By the end of the first week the mice receiving YK-4-279 had much lower numbers of leukemia cells. At the end of two weeks the treated mice were nearly normal by many measures, while the untreated mice had overwhelming numbers of cancer cells and died on average after three weeks, the researchers say. By contrast, mice receiving only two weeks of YK-4-279 lived nearly three times as long.
"The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can't be answered until we conduct clinical trials," Üren explains. "We are looking for ways that would allow us to administer more of it, or even to formulate a pill."
Üren says much more work remains for the team in order to translate this drug from a laboratory application into clinical trials.
Support for this work came from the Children's Cancer Foundation, St. Baldrick's Foundation, Go4theGoal, the Burroughs Wellcome Fund Clinical Scientist Award in Translational Research, the Austrian Science Fund (FWF), the Children´s Cancer Research Institute and from grants from the National Institutes of Health (RC4CA156509, R01CA133662, R01CA138212).








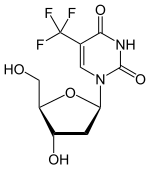 (Trifluridine)
(Trifluridine) 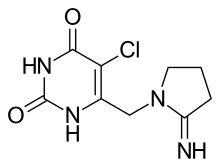 (Tipiracil)
(Tipiracil)


