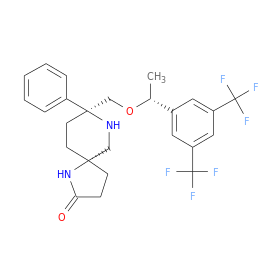
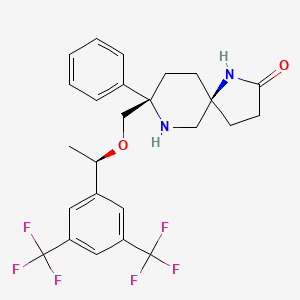
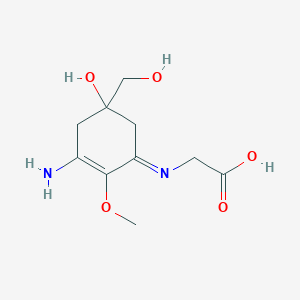
In continuation of my update on glycopyrrolate
Sunovion Pharmaceuticals Inc. (Sunovion) today announced that the U.S. Food and Drug Administration (FDA) approved the New Drug Application (NDA) for Lonhala Magnair (glycopyrrolate) Inhalation Solution (25 mcg twice daily), also known as SUN-101/eFlow®, for the long-term, maintenance treatment of airflow obstruction in people with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. Sunovion expects Lonhala Magnair to be available in U.S. pharmacies in early 2018.
Lonhala Magnair is the first nebulized long-acting muscarinic antagonist (LAMA) approved for the treatment of COPD in the U.S. and the first use of the Magnair, which is based on the closed eFlow® technology system, developed by PARI Pharma GmbH, to treat COPD. This technology is a virtually silent, portable, closed system nebulizer that is designed to deliver the drug in two to three minutes and allows people to breathe normally while using the device.
“We are proud that the FDA has approved Lonhala Magnair as the first nebulized, long-acting muscarinic antagonist treatment option for people in the U.S. living with COPD,” said David Frawley, Executive Vice President and Chief Commercial Officer at Sunovion. “The approval of Lonhala Magnair underscores our leadership in nebulization and the value we place on providing innovative treatment options for people living with COPD. Lonhala Magnair is an important addition to our portfolio of approved COPD therapies for people at various stages of COPD, providing the flexibility to choose handheld or nebulized products based on individual needs.”
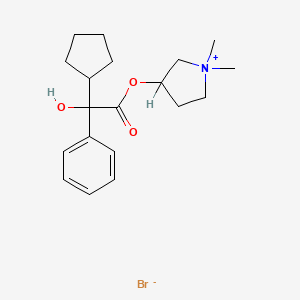
“Despite the availability of several therapies, many people still struggle to control their COPD – a challenge that may be affected by the delivery method used to administer a medication,” said Gary Ferguson, M.D., Pulmonary Research Institute of Southeast Michigan, Farmington Hills, Michigan. “Lonhala Magnair offers an important new option that combines the efficacy of a proven medication for COPD with the attributes of a unique handheld nebulizer that allows a person to breathe normally while taking their medication.”
Approximately 15.7 million adults in the U.S. report they have been diagnosed with COPD, a common, preventable and treatable disease that is characterized by persistent respiratory symptoms and airflow limitation that is due to airway and/or lung abnormalities usually caused by significant exposure to toxic particles or gases.1,2 The main risk factor for COPD is tobacco smoking, but other environmental exposures may contribute.2 The disease makes it hard for people to breathe and subsequently may limit their ability to perform some routine activities, including the proper inhalation of medication.2 This improper medication technique may impact treatment over time and may also result in an inadequate amount of the drug reaching the lungs, potentially worsening a person’s COPD.3,4,5 For people with moderate-to-very-severe COPD, nebulized treatments offer an alternative to inhalers, allowing a person to breathe normally while taking their medicine.
The approval is based on data from the clinical trials in the Glycopyrrolate for Obstructive Lung Disease via Electronic Nebulizer (GOLDEN) program, which included GOLDEN-3 and GOLDEN-4, two Phase 3, 12-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter, efficacy and safety trials comparing LONHALA MAGNAIR with placebo in adults with moderate-to-very-severe COPD. At study endpoints, individuals treated with Lonhala Magnair demonstrated statistically significant and clinically important changes from baseline in trough forced expiratory volume in one second (FEV1) at Week 12 versus placebo. An additional study, GOLDEN-5, was a Phase 3, 48-week, randomized, open-label, active-controlled, parallel-group, multicenter safety trial designed to evaluate the long-term safety and tolerability of Lonhala Magnair in adults with moderate-to-very-severe COPD and included the active comparator Spiriva® (tiotropium bromide) delivered by the HandiHaler® device. Lonhala Magnair was generally well-tolerated in clinical studies, with the most common side effects being exacerbations and cough. The overall treatment emergent adverse events (TEAE) incidences were similar for glycopyrrolate and tiotropium groups over 48 weeks.
 Chloroquine
Chloroquine  Hydroxy chlroquine
Hydroxy chlroquine