
Teva Pharmaceutical Industries Ltd. announced that it has received a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) regarding the New Drug Application (NDA) for SD-809 (deutetrabenazine) tablets for the treatment of chorea associated Huntington disease (HD). This is the first deuterated product to be reviewed by the FDA. The FDA has asked Teva to examine blood levels of certain metabolites. These metabolites are not novel, and are the same seen in subjects who take tetrabenazine or deutetrabenazine. No new clinical trials have been requested.
“Teva will continue to work closely with the FDA to bring SD-809 to the market as quickly as possible,” said Michael Hayden, M.D., Ph.D., President of Global R&D and Chief Scientific Officer at Teva. “We know that many people in the HD community are waiting for this new medicine. We understand there are very limited treatment options for HD patients and their families, hence we are accelerating the re-analysis process we were asked to conduct. We plan to submit our response to the CRL in Q3 2016."
HD is a rare and fatal neurodegenerative disorder caused by the death of nerve cells in the brain that affects about one in 7,000 – 10,000 people in western countries. Chorea—abnormal, involuntary writhing movements—is one of the most striking physical manifestations of this disease and it occurs in approximately 90% of patients at some point in the course of their illness.
In addition to HD, Teva’s programs for the development of SD-809 for the treatment of patients with tardive dyskinesia (TD) and Tourette syndrome (TS) are ongoing. Teva is currently conducting a Phase III efficacy and safety study in patients with moderate to severe TD known as AIM-TD (Addressing Involuntary Movements in Tardive Dyskinesia) and expects additional data from this study later in 2016, with regulatory submission to follow as planned. SD-809 has also been granted orphan drug designation for the treatment of TS in the pediatric population (defined as up to 16 years of age) and is planning further evaluation of SD-809 as a treatment for tics associated with TS.
About SD-809 (deutetrabenazine) Tablets
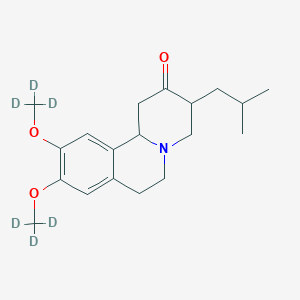
SD-809 (deutetrabenazine) is an investigational, oral, small-molecule inhibitor of vesicular monoamine 2 transporter, or VMAT2, that is being developed for the treatment of chorea associated with Huntington disease (HD). Deutetrabenazine has been granted Orphan Drug Designation for the treatment of HD by the U.S. Food and Drug Administration (FDA). Teva is also investigating the potential of deutetrabenazine for treating tardive dyskinesia, for which the FDA has granted a breakthrough therapy designation, and for tics associated with Tourette syndrome, for which the FDA has granted orphan status for pediatric use. Deutetrabenazine uses Teva’s deuterium technology.

 (Dapsone)
(Dapsone) Palbociclib
Palbociclib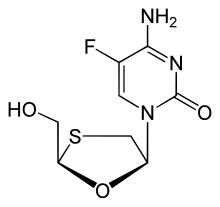 Emtricitabine
Emtricitabine  Rilpivirine
Rilpivirine Tenofovir alafenamide
Tenofovir alafenamide


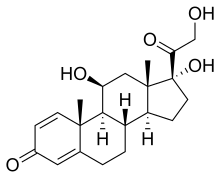 (Prednisole)
(Prednisole) (Indomethacin)
(Indomethacin)