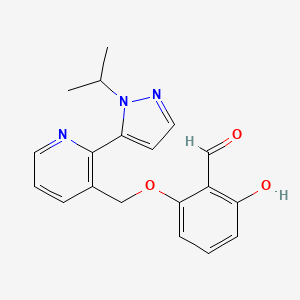
In continuation of my update on avatrombopag

Dova Pharmaceuticals, Inc. a pharmaceutical company focused on acquiring, developing and commercializing drug candidates for diseases where there is a high unmet need, today announced the U.S. Food and Drug Administration (FDA) approved a supplemental New Drug Application (sNDA) that expands the use of Doptelet (avatrombopag) to include the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment.
Doptelet is also FDA-approved for the treatment of thrombocytopenia in adult patients with chronic liver disease (CLD) who are scheduled to undergo a procedure. Earlier this week, Dova announced the marketing authorization granted by the European Commission for Doptelet for the treatment of severe thrombocytopenia in adult patients with CLD who are scheduled to undergo an invasive procedure.
“Dova is pleased to provide Doptelet to patients and physicians in the United States for the treatment of chronic ITP in adult patients who have had an insufficient response to a previous treatment,” said Dr. David Zaccardelli, president and CEO of Dova. “In addition to offering patients with ITP a new treatment option, we expect Doptelet will also address an important unmet medical need in the market. We sincerely thank the patients and dedicated researchers who participated in our clinical program as well as FDA for their collaboration during the review of this application.”
Doptelet is an oral, thrombopoietin receptor agonist (TPO-RA) administered with food. In the pivotal Phase 3 study, Doptelet administration resulted in a platelet count of at least 50,000 per µL at day eight of therapy in the majority of patients, with efficacy superior to placebo in maintaining platelet counts in the target range during the 6-month treatment period. Additional supportive efficacy data for the ITP sNDA were provided by two Phase 2 ITP clinical trials, as well as two Phase 3 trials for the treatment of thrombocytopenia in patients with CLD.
Safety data for 128 patients with ITP, and more than 1,000 subjects treated across 24 studies in the Doptelet clinical development program across multiple indications, support the safety and tolerability of Doptelet.
“ITP patients should work with their clinician to choose a therapy that supports their lifestyle and aims to achieve the best possible result to treat their ITP. That’s why having additional treatment options are so important,” said Caroline Kruse, president and CEO of the Platelet Disorder Support Association, a patient advocacy organization dedicated to ITP patients. “We are thrilled to have a new, oral TPO-RA available for adult patients with ITP. Every new treatment provides more choices and new hope to our community.”
Dova is committed to enabling patient access to Doptelet. Doptelet will be priced similarly to other TPO-RAs used to treat ITP, and Dova will continue to offer Patient Assistance and Co-Pay programs. The commercial launch of Doptelet for ITP is anticipated to occur in mid-July 2019.
Dova also entered into an expanded partnership in the United States with Salix. Starting on July 1, 2019, in addition to the gastroenterology, colorectal surgery, and proctology segments, Salix will have the exclusive right to co-promote the CLD indication for Doptelet to the hepatology and interventional radiology segments. Dova will continue to pay Salix a commission based on a percentage of net sales in these specialties, which will be in the mid-thirties beginning on July 1, 2019. In addition, the co-promotion agreement was extended to September 2023.
Dr. Zaccardelli added, “The expanded partnership with Salix builds additional momentum for Doptelet and enables the Dova team to focus on a successful launch of the ITP indication. As a growing leader in the treatment of thrombocytopenia, we are committed to realizing Doptelet’s significant market opportunity in CLD, ITP and potentially chemotherapy-induced thrombocytopenia (CIT) for which we expect Phase 3 trial top-line results in the first half of 2020.”
https://pubchem.ncbi.nlm.nih.gov/compound/AVATROMBOPAG#section=3D-Conformer
https://en.wikipedia.org/wiki/Avatrombopag