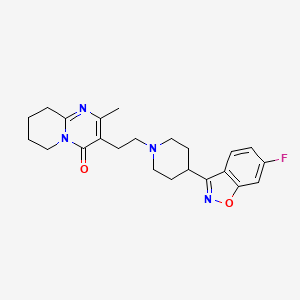
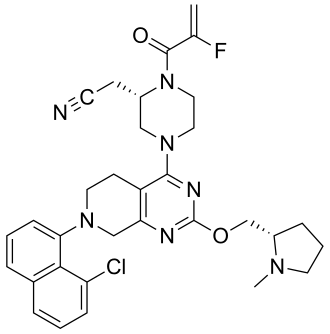
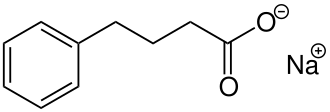
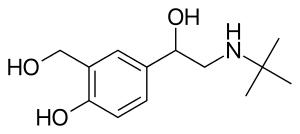
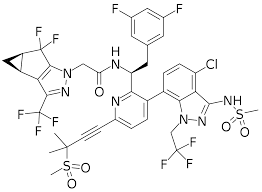
In continuation of my update on aripiprazole
Otsuka Pharmaceutical Co., Ltd. (Otsuka) and Lundbeck Pharmaceuticals LLC (Lundbeck) announced the U.S. Food and Drug Administration (FDA) approval of the New Drug Application (NDA) for Abilify Asimtufii® (aripiprazole) extended-release injectable suspension for intramuscular use, a once-every-two-months injection for the treatment of schizophrenia in adults or for maintenance monotherapy treatment of bipolar I disorder in adults.
Abilify Asimtufii offers two months of sustained therapeutic concentrations with one dose. Each dose is provided in a single-chamber, prefilled syringe, and is administered by a healthcare professional to appropriate patients via intramuscular injection in the gluteal muscle. Long-acting injectables provide continuous delivery of antipsychotic medication and can maintain therapeutic plasma concentrations, which may help to maintain symptom control of schizophrenia and bipolar I disorder.
"We are pleased to offer this new treatment option for people living with schizophrenia or bipolar I disorder that may delay the time to relapse," said John Kraus, M.D., Ph.D., executive vice president and chief medical officer at Otsuka Pharmaceutical Development & Commercialization, Inc. "This approval underscores Otsuka's commitment to innovate and continuously evolve to meet the needs of the communities we serve."
The efficacy of Abilify Asimtufii is based on the adequate and well-controlled studies of Abilify Maintena (aripiprazole) in the treatment of schizophrenia or maintenance treatment of bipolar I disorder in adults. The aripiprazole concentrations of Abilify Asimtufii were explored in a pharmacokinetic bridging study which was a 32-week, open-label, multiple-dose, randomized, parallel-arm, multicenter study (N=266) in patients living with schizophrenia and bipolar I disorder. The once-every-two-months, long-acting injectable formulation in 960 mg and 720 mg prefilled syringes delivers sustained plasma concentrations similar to that demonstrated in studies with aripiprazole monohydrate once-monthly, long-acting injectable, resulting in similar sustained efficacy.
"This approval is important news for patients, families, and healthcare providers. We hope that the use of Abilify Asimtufii in treatment plans will have a positive impact on those living with schizophrenia or bipolar I disorder," said Johan Luthman, executive vice president, R&D, Lundbeck. "We are grateful to the patients and researchers who made this major milestone possible."