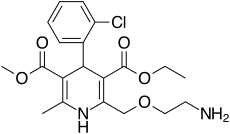
Dermavant Sciences, a biopharmaceutical company dedicated to developing and commercializing innovative therapeutics in immuno-dermatology, announced that the U.S. Food and Drug Administration (FDA) has approved Vtama (tapinarof) cream, 1%, an aryl hydrocarbon receptor agonist, indicated for the topical treatment of plaque psoriasis in adults. This approval makes Vtama cream the first and only FDA-approved steroid-free topical medication in its class.
“We are delighted with our FDA-approved label for Vtama cream, which is for adults with psoriasis, regardless of disease severity, and with an unlimited duration of use. In anticipation of today’s approval, we have a fully built commercial infrastructure in place, and I am excited to say we will have product in the channel in the first week of June. As the first and only approved drug in its class in the U.S., the FDA’s approval of Vtama cream provides an effective new non-steroidal treatment option for millions of adults living with plaque psoriasis and represents a major milestone for Dermavant and its stakeholders,” said Todd Zavodnick, Chief Executive Officer of Dermavant. “At Dermavant, we are committed to advancing novel, patient-focused innovation in immuno-dermatology. As such, we are proud to have developed a topical treatment in Vtama cream that provides not only efficacy over 52 weeks but can also be used on all body areas, including on sensitive locations, such as face, skin folds, neck, genitalia, anal crux, inflammatory areas, and axillae. In addition, an approximately four month off-treatment remittive effect (median time to first worsening), leads us to believe that Vtama cream has the potential to become the preferred topical option for this chronically underserved patient population and among the physicians who treat them.”
“Following 20-plus years of minimal innovation in the topical psoriasis treatment space1,2,3,4, I believe the approval of Vtama cream is an important step in establishing a new treatment option for adults with mild, moderate and severe plaque psoriasis ,” said Mark Lebwohl, MD, FAAD, Dean for Clinical Therapeutics and Waldman Professor and Chairman Emeritus of the Kimberly and Eric J. Waldman Department of Dermatology, Icahn School of Medicine at Mount Sinai in New York and lead author of the Phase 3 studies of Vtama cream published in The New England Journal of Medicine. “As a clinician, I’m excited to finally have a versatile, once-daily, steroid-free topical treatment that is backed by extensive clinical trial data supporting its favorable safety and efficacy profile and a demonstrated remittive effect of approximately four months in patients off therapy.”
Across PSOARING 1 and PSOARING 2, Vtama cream demonstrated highly statistically significant improvement in Physician Global Assessment (PGA)6 score of “clear” (PGA=0) or “almost clear” (PGA=1) with a minimum 2-grade improvement compared with vehicle from baseline at week 12. Vtama cream also demonstrated a highly statistically significant improvement in all secondary endpoints versus vehicle, including ≥75% Improvement in Psoriasis Area and Severity Index (PASI) score (PASI-75) from baseline at week 127. The adverse event (AE) profile of Vtama cream reported in both PSOARING 1 and PSOARING 2 demonstrated that the majority of AEs were localized to the site of application and were mild to moderate in nature. The most common AEs of subjects treated with Vtama cream were folliculitis, nasopharyngitis, and contact dermatitis.