Researchers at ETH Zurich and ETH spin-off Inositec have developed a new substance to prevent vascular calcification, which affects many patients suffering from chronic kidney disease. As their metabolism is impaired, calcium salts may deposit in soft tissues, such as blood vessels or even the heart valves, causing them to stiffen. This often leads to severe, potentially fatal cardiovascular diseases. However, before patients can benefit from the substance further research and tests must be carried out.

"Calcification occurs when calcium phosphate crystals are deposited in tissue," explains Jean-Christophe Leroux, professor of drug formulation and delivery at ETH Zurich. "The compound adheres to calcium phosphate crystals, inhibiting their growth."
Derivative of a natural substance
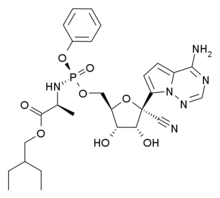
The new molecule is structurally related to inositol hexakisphosphate, also known as IP6. Occurring naturally in legumes and cereals, IP6 binds phosphate and various minerals, such as calcium, magnesium and iron. The plants use the molecule in their seeds to provide the seedlings with a sufficient supply of these substances.
It has been known for some time that IP6 also has an effect in the human bloodstream. The molecule has to be injected as it cannot be absorbed after oral ingestion. Other scientists are currently conducting clinical trials to study how effectively IP6 prevents vascular calcification.
Screening the collection of molecules
"The problem, though, is that IP6 is not particularly stable and is metabolized by the body very quickly," Antonia Schantl says. A doctoral student in Leroux's group, she is the lead author of the paper that has been published in the journal Nature Communications. In order to overcome this problem, Leroux and his colleagues sought to stabilize the molecule by making specific chemical modifications. They developed a series of related molecules, which ETH then patented. To be able to market one or more of these derivatives as medication in the future, ETH Professor Leroux and others involved founded the spin-off Inositec, which acquired the licence from ETH to use the molecule family.
Leroux's group at ETH subsequently collaborated with Inositec and researchers from other universities to screen this collection of molecules in a project that was co-financed by the Swiss innovation agency Innosuisse. The scientists conducted in vitro experiments to study the molecules' ability to inhibit the growth of calcium phosphate crystals in the blood and check their stability. They also tested their effect in a disease model in rats. The studies singled out one of the molecules in the collection as particularly suitable.
For the next stage, the ETH scientists will work with Inositec and third parties to clarify various issues, such as drug safety and the optimal dosage.
https://www.nature.com/articles/s41467-019-14091-4
https://en.wikipedia.org/wiki/Phytic_acid
https://en.wikipedia.org/wiki/Phytic_acid