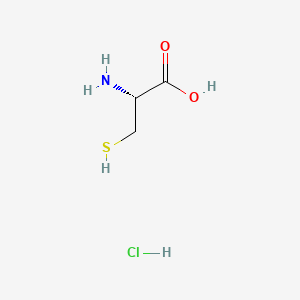
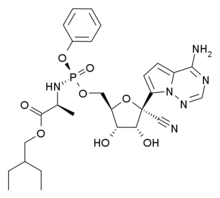
In continuation of my update on voxelotor

Global Blood Therapeutics, Inc. (GBT) (NASDAQ: GBT) announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval for Oxbryta (voxelotor) tablets for the treatment of sickle cell disease (SCD) in adults and children 12 years of age and older. Oxbryta, an oral therapy taken once daily, is the first approved treatment that directly inhibits sickle hemoglobin polymerization, the root cause of SCD. The medicine is expected to be available through GBT’s specialty pharmacy partner network within two weeks.
“Today is a major milestone not only for GBT but, most importantly, for people living with SCD, their families and those who care for them. When we started our journey with the SCD community more than eight years ago, we set out to transform the way this devastating, lifelong disease is treated,” said Ted W. Love, M.D., president and chief executive officer of GBT. “We are proud to bring this breakthrough therapy to the SCD community. Uniquely developed from inception to treat SCD, Oxbryta embodies GBT’s commitment to develop and deliver innovative medicines for patients with overlooked, life-limiting chronic diseases. We are grateful to the patients, caregivers, clinical trial investigators, healthcare providers and advocates who have worked alongside us to develop this first-in-class therapy.”
SCD affects an estimated 100,000 people in the United States and millions of people throughout the world, particularly among those whose ancestors are from sub-Saharan Africa. It also affects people of Hispanic, South Asian, Southern European and Middle Eastern ancestry.4 SCD is a lifelong inherited blood disorder that impacts hemoglobin, a protein carried by red blood cells that delivers oxygen to tissues and organs throughout the body.2 Due to a genetic mutation, people with SCD form abnormal hemoglobin known as sickle hemoglobin. Through a process called hemoglobin polymerization, red blood cells become sickled – deoxygenated, crescent-shaped and rigid.2,3,5 The sickling process causes hemolytic anemia (low hemoglobin due to red blood cell destruction) and blockages in capillaries and small blood vessels, which impede the flow of blood and oxygen throughout the body. The diminished oxygen delivery to tissues and organs can lead to life-threatening complications, including stroke and irreversible organ damage.
“Every person with SCD experiences hemoglobin polymerization and suffers from varying severity of anemia and hemolysis,” said Elliott Vichinsky, M.D., director of hematology/oncology at UCSF Benioff Children’s Hospital in Oakland, California. “With today’s approval of Oxbryta, we now have a therapy that significantly improves hemoglobin levels, has a favorable safety profile and reduces the anemia and hemolysis that inevitably leads to the long-term and often undetected detrimental effects associated with this chronic genetic condition.”
The accelerated approval of Oxbryta is based on clinically meaningful and statistically significant improvements in hemoglobin levels, accompanied by reductions in red blood cell destruction (hemolysis). Data from the Phase 3 HOPE (Hemoglobin Oxygen Affinity Modulation to Inhibit HbS PolymErization) Study of 274 patients 12 years of age and older with SCD showed that, after 24 weeks of treatment, 51.1% of patients receiving Oxbryta achieved a greater than 1 g/dL increase in hemoglobin compared with 6.5% receiving placebo (p<0.001). Results from the HOPE Study were published in June 2019 in The New England Journal of Medicine.
The most common adverse reactions occurring in ≥10% of patients treated with Oxbryta with a difference of >3% compared to placebo were headache (26% vs. 22%), diarrhea (20% vs. 10%), abdominal pain (19% vs. 13%), nausea (17% vs. 10%), fatigue (14% vs. 10%), rash (14% vs. 10%) and pyrexia (12% vs. 7%).
“SCD is a devastating, lifelong, inherited blood disorder that greatly impacts a person’s life, including their ability to work, attend school and look after their families, and it can reduce their overall life expectancy,” said Beverley Francis-Gibson, president and CEO of the Sickle Cell Disease Association of America. “After decades of waiting, we now have a treatment option that could change the course of this disease. We look forward to continuing to collaborate with GBT on initiatives aimed at transforming the care of patients living with SCD and ensuring access to important and innovative new medicines.”
GBT is committed to ensuring that people with SCD who are prescribed Oxbryta have help accessing the medicine. The company has established GBT Source™, a comprehensive program for patients who are prescribed Oxbryta that provides a wide range of practical, educational and financial support customized to each patient’s needs. GBT Source provides support by reviewing insurance coverage options and explaining benefits; working with the specialty pharmacy partner network to coordinate delivery of Oxbryta to wherever the patient chooses; helping with financial and co-pay assistance for eligible patients; and helping patients stay on treatment as prescribed by their treating physicians with a nurse support team. More information is available at www.Oxbryta.com or 1-833-428-4968 (1-833-GBT-4YOU).
The FDA instituted its accelerated approval pathway to allow for earlier approval of drugs that treat serious conditions and that fill an unmet medical need based on a surrogate endpoint.9 As a condition of accelerated approval, GBT will continue to study Oxbryta in the HOPE-KIDS 2 Study, a post-approval confirmatory study using transcranial doppler (TCD) flow velocity to demonstrate a decrease in stroke risk in children 2 to 15 years of age. The study will be initiated by the end of the year.
In recognition of the critical need for new SCD treatments, the FDA reviewed Oxbryta under Priority Review and granted Oxbryta Breakthrough Therapy, Fast Track, Orphan Drug and Rare Pediatric Disease designations for the treatment of patients with SCD.
Indication
Oxbryta is a prescription medicine used for the treatment of sickle cell disease in adults and children 12 years of age and older. It is not known if Oxbryta is safe and effective in children below 12 years of age.
This indication is approved under accelerated approval based on increase in hemoglobin (Hb). Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
Important Safety Information
Oxbryta should not be taken if the patient has had an allergic reaction to voxelotor or any of the ingredients in Oxbryta. See the end of the patient leaflet for a list of the ingredients in Oxbryta.
Oxbryta can cause serious side effects, including serious allergic reactions. Patients should tell their healthcare provider or get emergency medical help right away if they get rash, hives, shortness of breath or swelling of the face.
Patients receiving exchange transfusions should talk to their healthcare provider about possible difficulties with the interpretation of certain blood tests when taking Oxbryta.
The most common side effects of Oxbryta include headache, diarrhea, stomach (abdominal) pain, nausea, tiredness, rash and fever. These are not all the possible side effects of Oxbryta.
Before taking Oxbryta, patients should tell their healthcare provider about all medical conditions, including if they have liver problems; if they are pregnant or plan to become pregnant as it is not known if Oxbryta can harm an unborn baby; or if they are breastfeeding or plan to breastfeed as it is not known if Oxbryta can pass into breastmilk or if it can harm a baby. Patients should not breastfeed during treatment with Oxbryta and for at least 2 weeks after the last dose.
Patients should tell their healthcare provider about all the medicines they take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Some medicines may affect how Oxbryta works. Oxbryta may also affect how other medicines work.
https://en.wikipedia.org/wiki/Voxelotor