Gilead Sciences, Inc. (Nasdaq: GILD) announced that STELLAR-4, a Phase 3, randomized, double-blind, placebo-controlled study evaluating the safety and efficacy of selonsertib, an investigational, once-daily, oral inhibitor of apoptosis signal-regulating kinase 1 (ASK1), in patients with compensated cirrhosis (F4) due to nonalcoholic steatohepatitis (NASH), did not meet the pre-specified week 48 primary endpoint of a ≥ 1-stage histologic improvement in fibrosis without worsening of NASH.
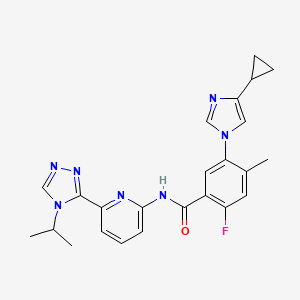
In the study of 877 enrolled patients who received study drug, 14.4 percent of patients treated with selonsertib 18 mg (p=0.56 vs. placebo) and 12.5 percent of patients treated with selonsertib 6 mg (p=1.00) achieved a ≥ 1-stage improvement in fibrosis according to the NASH Clinical Research Network (CRN) classification without worsening of NASH after 48 weeks of treatment, compared with 12.8 percent of patients who received placebo. Selonsertib was generally well-tolerated and safety results were consistent with prior studies.
“While we are disappointed that the STELLAR-4 study did not achieve its primary endpoint, we remain committed to advancing therapies for patients with advanced fibrosis due to NASH, where there is a significant unmet need for effective and well-tolerated treatments. Gilead has a long-term commitment and proven track record of addressing significant challenges in the field of liver diseases. Data from this large study of patients with compensated cirrhosis due to NASH, including the extensive set of biomarkers collected, will further advance our understanding of the disease and inform our broader NASH development programs,” said John McHutchison, AO, MD, Chief Scientific Officer, Head of Research and Development, Gilead. “We are grateful to the patients and investigators who participated in the STELLAR-4 study, and we now await the upcoming results from the Phase 3 STELLAR-3 trial of selonsertib in patients with bridging fibrosis (F3) due to NASH and the Phase 2 ATLAS combination trial of selonsertib, cilofexor (GS-9674) and firsocostat (GS-0976) in patients with advanced fibrosis due to NASH later this year.”
Further in-depth analysis of the findings is ongoing and the data will be submitted to an upcoming scientific conference. Gilead will work with the Data Monitoring Committee and investigators to conclude the STELLAR-4 study in a manner consistent with the best interests of each patient.
Selonsertib, cilofexor and firsocostat, alone or in combination, are investigational compounds and are not approved by the U.S. Food & Drug Administration (FDA) or any other regulatory authority. Safety and efficacy have not been established for these agents.
About Selonsertib and the STELLAR-4 Study
Selonsertib is an investigational small molecule inhibitor of ASK1, a protein that promotes inflammation, apoptosis (cell death) and fibrosis in settings of oxidative stress. Oxidative stress can be increased in many pathological conditions including liver diseases such as NASH.
The STELLAR-4 study is a Phase 3, randomized, double-blind, placebo-controlled study evaluating the safety and efficacy of selonsertib in patients with compensated cirrhosis (F4) due to NASH. Eligible adults ages 18 to 70 years were randomized and received selonsertib 18 mg (n=354), selonsertib 6 mg (n=351) or placebo (n=172) for up to 240 weeks. Either selonsertib or placebo is being administered orally once daily. The primary endpoints of the study are a composite of the proportion of patients who achieve a ≥ 1-stage improvement in fibrosis according to the NASH CRN classification without worsening of NASH at week 48 and event-free survival at week 240 as assessed by time to the first clinical event. Further information about the clinical study can be found at www.clinicaltrials.gov.
https://pubchem.ncbi.nlm.nih.gov/compound/Selonsertib#section=2D-Structure