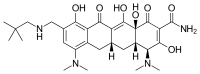
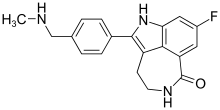
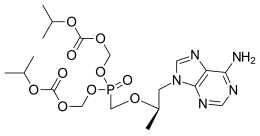
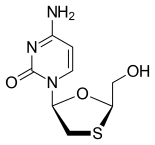
In continuation of my update on zoledronic acid
Researchers in China have discovered that an enzyme called UGT8 drives the progression of basal-like breast cancer, an aggressive form of the disease that is largely untreatable. But the study, which will be published May 4 in the Journal of Experimental Medicine, reveals that the widely used osteoporosis drug zoledronic acid inhibits UGT8 and prevents the spread of basal-like breast cancer in mice, suggesting that this drug could also be used to treat the disease in humans.
Basal-like breast cancer is a particularly aggressive form of breast cancer that typically affects younger, premenopausal women. It is hard to treat because the tumor cells are usually "triple negative," lacking the estrogen receptor, progesterone receptor, and HER2 protein that are the main therapeutic targets for other forms of breast cancer. As a result, the prognosis for patients with basal-like breast cancer is worse than any other breast cancer subtype.
"The highly aggressive nature and the absence of effective therapeutics for basal-like breast cancer make it a high priority to elucidate what determines its aggressiveness and identify potential therapeutic targets," says Professor Chenfang Dong from the Zhejiang University School of Medicine in Hangzhou, China.
Cancer cells must alter their metabolism in order to survive and metastasize to other parts of the body. Dong and colleagues examined over 5,000 breast cancer patient samples and found that levels of the metabolic enzyme UGT8 were dramatically elevated in patients with basal-like breast cancer. Higher UGT8 levels correlated with increased tumor size, higher tumor grade, and shorter patient survival times.
UGT8 catalyzes the first step in the synthesis of sulfatide, a type of lipid that is found on the surface of cells and has been implicated in cancer progression. Dong and colleagues found that breast cancer cells expressing high levels of UGT8 produce large amounts of sulfatide, which in turn activates signaling pathways crucial for the survival and metastasis of basal-like breast cancer. Depleting UGT8 from these cells lowered sulfatide levels and reduced the cells' ability to form tumors when injected into mice.
Zoledronic acid is a drug that is approved to treat a variety of bone diseases, including osteoporosis, and is on the World Health Organization's list of safe and effective medicines essential for global health. Dong and colleagues confirmed that zoledronic acid is a direct inhibitor of UGT8 that reduces the levels of sulfatide in basal-like breast cancer cells. Treatment with the drug impaired the cells' ability to invade their surroundings and, accordingly, prevented them from metastasizing to the lungs after they were injected into the mice.
"Our study suggests that UGT8 contributes to the aggressiveness of basal-like breast cancer and that pharmacological inhibition of UGT8 by zoledronic acid offers a promising opportunity for the clinical treatment of this challenging disease," Dong says.
Ref :http://jem.rupress.org/content/early/2018/05/03/jem.20172048
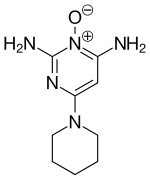 Finasterite
Finasterite 
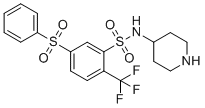 WAY-316606
WAY-316606