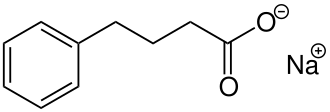
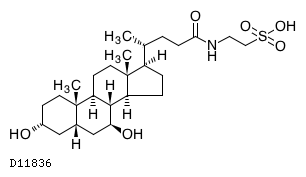
In continuation of my update on glycerol phenyl butyrate
Horizon Pharma plc (NASDAQ: HZNP) announced the U.S. Food and Drug Administration (FDA) has approved a supplemental new drug application (sNDA) to expand the age range for Ravicti (glycerol phenylbutyrate) Oral Liquid to include infants younger than two months of age living with a urea cycle disorder (UCD).
Ravicti is now FDA-approved for use as a nitrogen-binding agent for chronic management of UCDs in adults and children of all ages who cannot be managed by dietary protein restriction and/or amino acid supplementation alone. Ravicti must be used with dietary protein restriction and, in some cases, dietary supplements. Ravicti is not indicated for treatment of acute hyperammonemia in patients with UCDs, and its safety and efficacy for the treatment of n-acetylglutamate synthase (NAGS) deficiency has not been established.
“The FDA approval of Ravicti for children younger than two months provides a new alternative for the management of patients with a UCD that is easy to dose and administer to infants given the liquid formulation,” said Nicola Longo, M.D., Ph.D., clinical geneticist at Primary Children’s Hospital and the University of Utah Hospital, and a lead investigator of a clinical study evaluating Ravicti for newborns. “UCDs are severe and can be life-threatening. We hope that the combination of early diagnosis – through newborn screening or by measurement of ammonia levels – and the availability of novel treatments, such as this one, can help to improve the outcome of affected patients.”
A study was conducted to assess safety, efficacy and pharmacokinetics in pediatric patients with UCDs two months of age and younger (n=16). In the study, 10 patients transitioned to Ravicti from sodium phenylbutyrate, three transitioned from intravenous sodium benzoate and sodium phenylacetate, and three were treatment naïve. Patients were treated with Ravicti for an average of 10.7 months. Results demonstrated safety and efficacy in children younger than two months, with Ravicti-treated patients maintaining stable ammonia levels relative to their pre-study enrollment. In addition, mean ammonia levels were lower during treatment with Ravicti compared to baseline values.
“As we increase our efforts to develop new investigational medicines for people living with rare and rheumatic diseases, Horizon continues to seek ways to better serve patients with our current medicines,” said Elizabeth Thompson, Ph.D., vice president, clinical development, rare diseases, Horizon Pharma. “The FDA approval of Ravicti for children under the age of two months is a milestone in our efforts to help people living with UCDs, and we are proud to be bringing a new treatment option to the vulnerable newborn patient population.”
A UCD is a rare genetic disorder that affects approximately 1 in 35,000 live births in the United States. It is caused by an enzyme deficiency in the urea cycle, a process that is responsible for converting excess ammonia from the bloodstream and ultimately removing it from the body. Because of this, people with a UCD experience hyperammonemia, or elevated ammonia levels in their blood, that can then reach the brain and cause irreversible brain damage, coma or death. UCD symptoms may first occur at any age depending on the severity of the disorder, with more severe defects presenting earlier in life.
--------------------------------------------------------------------------------------------------------------------------