
Brain tissue can die as the result of stroke, traumatic brain injury, or neurodegenerative disease. When the affected area includes the motor cortex, impairment of the fine motor control of the hand can result. In a new study published in Restorative Neurology and Neuroscience, researchers found that inosine, a naturally occurring purine nucleoside that is released by cells in response to metabolic stress, can help to restore motor control after brain injury.
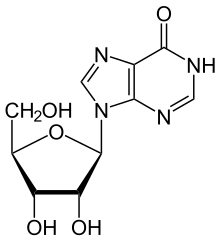
Based on evidence from rodent studies, researchers used eight rhesus monkeys ranging in age from 5 to 10 years (approximately equivalent to humans from 15 to 30 years of age). All received medical examinations and motor skills were tested, including video recording of fine motor functions used to retrieve small food rewards. All monkeys were given initial MRI scans to ensure there were no hidden brain abnormalities.
Brain injuries were created in the area controlling each monkey's favored hand. Four monkeys received inosine treatment, while four received a placebo. Research staff were not informed regarding which monkeys were included in the treatment vs placebo groups. Recovery of motor function was then measured for a period of 14 weeks after surgery.
While both the treated and placebo groups recovered significant function, three out of four of the treated monkeys were able to return to their pre-operative grasping methods. The placebo group developed a compensatory grasping method for retrieving food rewards unlike the original thumb-and-finger method.
"In the clinical context, the enhanced recovery of grasp pattern suggests that inosine facilitates greater recovery from this type of cortical injury and motor impairment," explained lead investigator Tara L. Moore, PhD, of the Department of Anatomy & Neurobiology and the Department of Neurology, Boston University School of Medicine, Boston, MA, USA. "To our knowledge, this is the first study to demonstrate the positive effects of inosine for promoting recovery of function following cortical injury in a non-human primate."
Inosine has also been administered in human clinical trials for multiple sclerosis and Parkinson's disease and has been proven to be safe in doses up 3000 mg/day. Athletes have used inosine as a nutritional supplement for decades, and inosine supplements are widely available commercially. "Given the effectiveness of inosine in promoting cortical plasticity, axonal sprouting, and dendritic branching, the present evidence of efficacy after cortical injury in a non-human primate, combined with a long history of safe use, indicates a need for clinical trials with inosine after cortical injury and spinal cord injury," noted Dr. Moore.
The study points to neural plasticity, whereby the brain essentially "re-wires" connections between neurons to reestablish control pathways, as a therapeutic target for the recovery of fine motor control and grasping ability. Further study of cortical tissue from these monkeys is currently being completed and may provide further insights into the mechanisms underlying recovery.


 Metformin
Metformin